Large-scale Genotyping and Genetic Mapping in Plasmodium Parasites
Article information
Abstract
The completion of many malaria parasite genomes provides great opportunities for genomewide characterization of gene expression and high-throughput genotyping. Substantial progress in malaria genomics and genotyping has been made recently, particularly the development of various microarray platforms for large-scale characterization of the Plasmodium falciparum genome. Microarray has been used for gene expression analysis, detection of single nucleotide polymorphism (SNP) and copy number variation (CNV), characterization of chromatin modifications, and other applications. Here we discuss some recent advances in genetic mapping and genomic studies of malaria parasites, focusing on the use of high-throughput arrays for the detection of SNP and CNV in the P. falciparum genome. Strategies for genetic mapping of malaria traits are also discussed.
INTRODUCTION
Human malaria is a deadly disease that still kills more than a million people each year, mostly in tropical and subtropical regions [1,2]. The human disease is caused by infection with 1 of the 5 Plasmodium parasites, Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malariae, and Plasmodium knowlesi. P. falciparum is the most deadly parasite, whereas P. vivax is the most wide-spread species. Although P. vivax is generally regarded as non-lethal, it can cause severe diseases in patients [3,4]. There are also malaria parasites that can infect many species of mammals, birds, and reptiles [5]. These animal malaria parasites can be excellent models for studying disease mechanisms and molecular pathways in the parasites.
The malaria parasite has a complex life cycle involving multiple rounds of sexual and asexual reproduction in its hosts. Sexual reproduction and genetic recombination of malaria parasites occur in mosquito vectors. The sexual stages are essential for transmission and genetic recombination that is critical for mapping studies. Most of the life cycle of a malaria parasite has a haploid genome. The genomes of the majority of malaria parasites are similar in size and contain 14 chromosomes with ~23-26 million nucleotides [6] and approximately 5,500 genes [7,8]. The nucleotide content (the percentage of G/A/T/C) can be very different: the P. falciparum genome has > 80% AT, whereas P. vivax has a much lower AT content (~63%) [7,9,10]. Another obvious difference is the species-specific gene families that encode variant antigens [6]. Unfortunately, the majority (60%) of the predicted genes in P. falciparum encode hypothetical proteins [7]. In this brief summary of malaria genetics and genomics, we shall focus on data from the P. falciparum parasite, although some information from other malaria parasites will also be briefly mentioned.
POLYMORPHISMS AND GENETIC MARKERS
A genetic mapping project generally consists of 3 parts: accurate measurement of phenotype, determination of genotype, and linkage or association analyses. Before a genetic mapping study can be performed, genetic markers for genotyping parasite DNA have to be developed. In the 1970s, polymorphisms in enzymes such as 6-replace with phosphogluconate hydrogenase and glucose-6-phosphate isomerase in starch gel electrophoresis were used to distinguish rodent malaria parasites and progeny from genetic crosses [11-13]. Restriction fragment-length polymorphisms (RFLP) became popular genetic markers for mapping malaria traits in the 1980s [14]. In the 1990s, microsatellites (MS) were introduced to fine map the chloroquine (CQ)-resistant gene [15, 16]. Now, in the 2000s, single-nucleotide polymorphism (SNP) is becoming the marker of choice because of the development of high-throughput SNP genotyping methods [17,18]. SNP genotyping is actually based on the same polymorphism as RFLP (nucleotide substitution); the difference is in the method for detecting the polymorphism. Large numbers of MS and SNP have been developed for P. falciparum [19-22]; and genetic markers for other parasites such as P. vivax and rodent malaria parasites are also available or being developed [23-27].
MS markers have advantages for population studies because they are highly polymorphic and generally more selectively neutral than many SNP [28]. Compared with SNP, the high mutation rates and multiple alleles of MS markers provide better resolving power for studying closely related parasite populations or for fine mapping loci with candidate genes. MS markers are therefore useful for parasite identification that generally does not require typing large numbers of markers. Multiple alleles of a MS marker make it valuable for searching signatures of drug selection within and between parasite populations [29-31]. The distribution and frequency of MS among different malaria parasite genomes are very different. MS are abundant in P. falciparum [7,15], mostly likely due to its high-AT-content genome, but relatively few are present in P. vivax [24] and P. yoelii [26].
SNP are also quite abundant in both P. falciparum and P. vivax genomes, although the distribution of SNP can vary greatly among various chromosomal regions or different genes [20-22,24,32,33]. In an earlier study of 204 genes from chromosome 3 of P. falciparum, approximately 1 SNP was found from about 900 bp DNA sequences among 5 P. falciparum isolates (π = 4.9 × 10-4) [32]. Recent large-scale sequencing suggested that the frequency of SNP in P. falciparum could be much higher [20,22]. High frequencies of SNP were found in genes encoding surface proteins [33] and putative transporters [34] that are likely under pressure from host immunity or antimalarial agents; but for housekeeping genes, the frequency of SNP can be much lower [35,36]. The uneven distribution of polymorphisms is one of the issues leading to the debate on the age of P. falciparum [32, 37,38]. In P. vivax, the frequency of SNP is also quite high, with approximately 1 SNP per 530 bp (π = 8.7 × 10-4) [24]. The frequency of nucleotide substitution, in addition to the abundance of polymorphic MS, makes it feasible to develop a high-resolution genetic map for malaria parasites, particularly P. falciparum.
PARASITE GENOTYPING METHODS
Over the years, various genetic typing methods have been developed. Polymorphisms in antigen-coding genes such as apical membrane antigen-1 (AMA-1), merozoite surface protein-1 (MSP-1), and circumsporozoite protein (CSP) have been widely used to type DNA samples collected from patients. More recently, MS markers are used because they are more likely to be neutral markers [15,39,40]. For typing and identification of parasite clones adapted to in vitro culture, genetic typing methods using multicopy genes can be very helpful [41-43]. Use of a selected number of MS markers (for example, 10 MS markers) or a set of SNP can be a very efficient and effective method for parasite 'fingerprinting' or parasite verification, as well [44-46].
MS products after PCR amplification can be labeled with radioactive or fluorescent materials and detected after separation in a DNA sequencing gel and an automatic DNA sequencer [44]. Because DNA sequencers are widely available, labeling PCR products with fluorescent dye is more convenient than labeling with isotopes; however, fluorescent-labeled primers can be expensive if large numbers of primers are required for a study; and a method using a single labeled universal primer for all the MS markers in a study can greatly reduce the primer cost [26]. Agarose gel can sometimes be used to resolve MS markers that have large size differences (4 bp or longer).
For high-throughput genotyping, it is clear that SNP is the marker of choice, because various high-throughput SNP typing methods are now available [47-52]. In P. falciparum, high-density arrays have been used to study genetic recombination in a progeny clone from the Dd2 × HB3 cross and to identify single-feature polymorphisms (SFPs) among field isolates [36,53]. Using an Affymetrix array with 298,782 25-mer P. falciparum probes, Kidgell et al. [36] identified 23,653 SFP and various copy-number variations (CNV) from the isolates. Similarly, genome-wide amplifications and deletions were also detected using oligonucleotides printed in glass slides [54]. More recently, another Affymetrix array containing ~2.5 million probes (PFSANGER array; designed at the Sanger Center, UK) became commercially available and has been used to study RNA transcripts [55].
In our laboratory, we have explored the possibility of using the Sanger tiling array for typing the P. falciparum genome. Biotin-labeled genomic DNA (10 µg) from a parasite was hybridized to the PFSANGER array at 45℃ for 16 hr, and signals from field isolates were compared with those from 3D7. If there is a difference (substitution) in a probe sequence between 3D7 and another parasite, signal from the parasite will be reduced compared with that of 3D7. Because hybridization signals can be influenced by various factors such as the position of a base substitution in a probe and the GC content of a probe, criteria used to distinguish a real substitution in a probe from background signals require extensive evaluation and verification of hybridization signals from different parasites. Our data showed that the Sanger array could be successfully used to detect polymorphism within a probe (SFP) with an accuracy rate higher than 90% after comparison of more than 3,000 known SNP [21] with those detected using the array [53]. We also showed that the last 2 end positions in a probe did not significantly affect hybridization signals and that probes with GC contents < 16% should be excluded from SFP calling because of poor hybridization signals. SFP calls based on a single probe were not reliable either; and a conservative signal cutoff ratio of 3-5.0 and a vote among several adjacent probes (within 25 bp) with a majority of the probes calling for an SFP (or mSFP for SFP called by multiple probes with 75% of the probes calling for polymorphism) should be applied. To illustrate how well the array performed in calling nucleotide polymorphism, we applied the SFP calling parameters to identify 5 known substitutions within a gene encoding a homolog of the human P-glycoprotein (pfmdr-1) [21,56]. We were able to identify 3 of the 5 known mutations (NIAID SNP ID: PFE1150w-1, PFE1150w-3, and PFE1150w-5) in the pfmdr-1 (Fig. 1A). The other 2 mutations (PFE1150w-2 and PFE1150w-4) were not detected because of absence of probes covering the substitution sites (Fig. 1B, C). Additionally, this array was a useful tool for detecting CNV in the P. falciparum genome. Genomic regions that were highly polymorphic, deleted, or amplified could be detected after comparison of hybridization signals from parasite isolates (Fig. 2). In particular, comparison of the chromosome 5 sequence from parasite 106/1 with that of 3D7 showed amplification of a ~100-kb segment containing pfmdr-1 in the 106/1 parasite (Fig. 2A). In addition, potential amplifications on chromosome 1, 2, 4, 8, and 13 and highly polymorphic var clusters could also be detected (Fig. 2B). CNV has received more attention lately because it has been shown to be associated with parasite response to antimalarial drugs [57-60]. The results from this study showed that the Sanger array could be used to call both SFP and CNV in the P. falciparum genome.
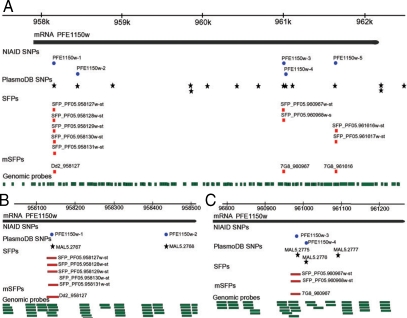
Genome browser displays (drawn in Canvas) showing SFP, mSFP and SNP, and probes at the pfmdr-1 locus. (A) A genome browser window (~5 kb) showing chromosomal region covering pfmdr-1 gene (top line) and the predicted pfmdr-1 coding region (black bar); blue circle, known SNPs reported previously [21,56], black stars, SNPs in PlasmoDB; red squares, SFPs detected by individual probe and mSFP by a majority of the probes; green bars, genomic probe covering the pfmdr-1 gene. Names of the SNPs and probes are as labeled. Note the mSFP matched well with those known SNPs. (B and C) An expanded view (500-bp window) showing 2 regions in the pfmdr-1, including 2 known substitutions (PFE1150w-1 and PFE1150w-4) that were not covered by any of the genomic probes. The labels are the same as in (A). For more information on genome-wide SNPs and SFPs, please visit: http://p-falcip.abcc.ncifcrf.gov/cgi-bin/gbrowse/malaria_sanger/.
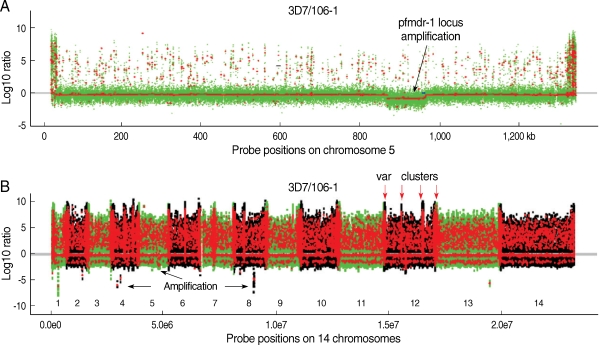
Chromosome segmentation analysis showing genomic amplification and deletion/highly polymorphic regions on chromosomes of Plasmodium falciparum. Signals from DNA hybridizations to the Sanger tiling microarray were normalized as described [53]. Chromosomal segments (seen as dots) from circular binary segmentation (CBS) analysis [91] of the data for 106/1 strain using 3D7 strain as reference were plotted. (A) Probe signal ratios (3D7 over 106/1) showing an amplification of a locus containing pfmdr-1 on chromosome 5 in the 106/1 parasite. Green dots are normalized log ratios of individual probe, and the red lines/dots are the mean values of normalized log ratios of all the probes in segments obtained after CBS analysis. Dots above the grey/red line at zero are polymorphic individual probes (green) or segments with multi-probes with similar reduced signals in 106/1 (red); and those under the line are potentially amplified regions (red). (B) CBS analysis applied to all chromosomes (1-14) of the 106/1 parasite using 3D7 as reference. The green dots are normalized log ratios of individual probes; and the red lines/dots are the mean values of normalized log ratios from all the probes in segments obtained after CBS analysis. The dots with positive values suggested polymorphisms between 106/1 and 3D7; and those with negative values were potential genomic amplification. Highly polymorphic chromosome ends and internal var gene clusters can be seen as intense red 'zones'. Red arrows point to 4 known var clusters on chromosome 12; and black arrows indicate some potential amplifications on chromosome 4, 5, and 8. The numbers 1-14 marks the parasite 14 chromosomes.
A P. falciparum-specific SNP typing array using the molecular inversion probe (MIP) method [61] has also been developed for typing parasite SNP in our lab (J. Mu et al., unpublished). Because genomic DNA is amplified using a pair of primers specific for an SNP to be assayed, the MIP method can detect SNP using relatively small amount of DNA (~100 ng). This method therefore can potentially be used for genotyping DNA samples collected directly from patients. If a DNA sample is amplified using commercial kits such as REPLI-g® Whole Genome Amplification (QIAGEN) that can increase the amount of DNA in a sample 300-400 times, even smaller amounts of DNA will be required. Before genome-wide amplification, however, human DNA has to be removed before DNA purification, because the human genome is ~100 times that of a malaria parasite genome. Many other genotyping arrays such as Affymetrix 3 K and 75 K arrays (S. Volkman, D. Wirth et al., Harvard University), NimbleGen 60-mer oligo array (M. Ferdig, Notre Dame University), and another Affymetrix tiling with ~5 million probes (E. Winzeler, Scripps) are being developed and will be available soon. These arrays and methods will greatly facilitate our ability to detect large numbers of genome polymorphisms in P. falciparum parasites.
MAPPING APPROACHES
Genetic mapping in malaria parasites can be generally classified into 3 different approaches. The first is linkage mapping using genetic crosses. The first genetic cross in malaria parasites was performed by Dr. David Walliker and his colleagues at the University of Edinburgh, UK [62]. Following the initial cross, many more genetic crosses have been performed in the human malaria parasite P. falciparum and in rodent malaria parasites [12-14,63-65]. One successful story of mapping malaria traits using progeny from a genetic cross was the identification of pfcrt, the gene that plays a key role in CQ resistance in P. falciparum [14,16,66,67]. The advantage of mapping using progeny from genetic crosses is that the genetic backgrounds in the progeny derive from the 2 parents, reducing genomic background noises seen in field isolates; however, performing a genetic cross can be expensive and laborious, not to mention the ethical concerns of using nonhuman primates for crosses of human malaria parasites. In this regard, use of progeny from crosses of rodent malaria parasites can be good alternatives. Although phenotypes in rodent malaria do not always exactly reflect those seen in human malaria parasites, what we learn from rodent malaria parasites will provide important information for studying human malaria parasites. Importantly, disease phenotypes such as virulence can be dissected more easily using rodent malaria parasites, because host genetic background variation can be controlled. The second approach is to investigate the association of candidate genes such as pfmdr-1, pfdhfr, and pfcrt with drug resistance in field isolates [34,68], but candidate genes have to be available or identified before a study can be conducted. The third approach is genome-wide association, which is becoming popular with the development of various high throughput genotyping methods [18,31]. The majority of genetic studies in malaria parasites have been performed using rodent malaria parasites (Plasmodium chabaudi, Plasmodium yoelii, Plasmodium berghei) and P. falciparum and have focused on mapping genes contributing to drug resistance [14,18,69-75]. Differences in parasite growth and development, virulence, transmission, immunogenicity, variation in gene expression, and a 'mutator' phenotype are some of the traits that have also been studied [76-83].
Genetic recombination (or crosses) occurs in the field when parasites are ingested by a mosquito; however, a recombination event will be detected only if 2 or more parasites with different genotypes are present. It is quite common for a mosquito to carry parasites with different genotypes, particularly in regions with relatively high transmission rates, such as Africa and Asia [39,84,85]. Genetic recombination occurring in parasite populations can be explored for mapping genes contributing to drug resistance and other parasite traits. Under drug pressure, genetic changes will occur in some parasites, leading to parasites that can survive at a high drug concentration. Some wild type parasites may acquire the changes through genetic recombination and survive. This process will select for parasites carrying drug-resistant mutations, leading to a drug 'selective sweep' that can be detected through genotyping. Signature of drug selection has been reported for CQ and sulfadoxine-pyrimethamine resistance [29,30,86-89].
We are interested in mapping genes that may affect parasite responses to various antimalarial drugs including quinine, mefloquine, and others. While developing high-throughput methods to genotype P. falciparum parasites collected from different regions of the world, we have also been testing the parasite responses to different drugs. Hundreds of parasites have been collected, adapted to in vitro culture, and tested. We insisted on testing parasite drug responses after adapting parasites from patients into in vitro culture and genotyping them to make sure they were clonal, because mixed infections and many other factors might influence drug test results [90]. Accurate measurement of drug response is particularly important when a phenotype being studied is controlled by multiple genes. DNA samples from the parasites are being typed using the MIP and the Sanger tiling microarrays. We hope to associate some polymorphisms in the parasite genome to elevated parasite responses (increased IC50) to various antimalarial drugs. After identification of candidate genes, molecular and biochemical approaches will be employed to evaluate the functions of the candidate genes.
CONCLUSION
High-throughput genotyping methods are now available for typing DNA from P. falciparum and for mapping parasite traits; and many more typing methods are under development, including those for other malaria species. Unfortunately, the malaria parasite is a single-cell organism, and it is thus challenging to detect or measure reproducible phenotypic variation between individual parasites. The future direction for mapping malaria traits should focus on developing methods to accurately characterize and measure phenotypic variation among individual parasites.
ACKNOWLEDGEMENTS
We thank Jun Yang and Brandie Fullmer at the Laboratory of Immunopathogenesis and Bioinformatics, SAIC-Frederick, Inc. for microarray hybridizations and NIAID intramural editor Brenda Rae Marshall for assistance. This work was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health and the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health; and in part was funded by NCI contract N01-CO-12400.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U. S. Government.
Because X-z.S., H.J., and J.M. are government employees and this is a government work, the work is in the public domain in the United States. Notwithstanding any other agreements, the NIH reserves the right to provide the work to PubMedCentral for display and use by the public, and PubMedCentral may tag or modify the work consistent with its customary practices. You can establish rights outside of the U.S. subject to a government use license.