PCR Diagnosis of Entamoeba histolytica Cysts in Stool Samples
Article information
Abstract
Amebiasis is a protozoan disease caused by Entamoeba histolytica and a potential health threat in areas where sanitation and hygiene are inappropriate. Highly sensitive PCR methods for detection of E. histolytica in clinical and environmental samples are extremely useful to control amebiasis and to promote public health. The present study compared several primer sets for small subunit (SSU) rDNA and histone genes of E. histolytica cysts. A 246 bp of the SSU rDNA gene of pure cysts contained in phosphate-buffered saline (PBS) and in stool samples was successfully amplified by nested PCR, using the 1,147-246 bp primer set, of the primary PCR products which were pre-amplified using the 1,147 bp primer as the template. The detection limit of the nested PCR using the 1,147-246 primer set was 10 cysts in both groups (PBS and stool samples). The PCR to detect histone gene showed negative results. We propose that the nested PCR technique to detect SSU rDNA can be used as a highly sensitive genetic method to detect E. histolytica cysts in stool samples.
Parasitic infections are endemic and represent a major public health problem worldwide. In particular, Entamoeba histolytica, a pathogenic protozoan that parasitizes the human small intestine and causes amebiasis, results in human infections on a global scale [1]. It has been reported that 40-50 million people worldwide develop E. histolytica-related dysentery or liver abscess, and as many as 100,000 people die of amebiasis every year [2]. The conventional method of diagnosing E. histolytica utilizes microscopy, but its sensitivity is less than 60% [3]. To improve the rate and sensitivity of the diagnosis, PCR-based approaches have been used since the early 1990s, and many studies have been conducted to detect target E. histolytica DNA in the human stool [4-7]. The small subunit rRNA (SSU rDNA) gene has been widely targeted to diagnose E. histolytica and can distinguish it from other ameba species [8,9]. Here, we describe a new nested PCR method to specifically and sensitively detect E. histolytica in human stool samples by targeting the SSU rRNA gene.
The E. histolytica HM-1 (IMSS c16) strain used in this study was kindly provided by Prof. Myeong-Heon Shin of Yonsei University, the Republic of Korea. E. histolytica cysts were counted using a hemocytometer, and 0 to 105 cysts were added to stool samples. DNA was extracted from the stool samples using a modified protocol of the formalin-ether method of Garcia and coworkers [10,11]. A stool sample, 1 g, was placed in a sterilized plastic cup, and 10 ml of distilled water was added. The solution was filtered once through gauze and centrifuged at 1,660 g for 10 min. The supernatant was removed, and 10 ml of 10% ethanol was added to the precipitate, which was completely dissolved using a disposable loop. Ether (3 ml) was added to the solution, which was shaken vigorously and centrifuged at 1,660 g for 10 min. The supernatant was completely removed, and DNA was extracted from the precipitate using a QIAamp DNA stool mini kit (Qiagen, Hilden, Germany). After centrifugation, the pellet precipitate was transferred to a 2 ml Eppendorf tube and completely dissolved in 1.4 ml of the ASL buffer from the kit. The solution was subjected to 5 times' freeze-thaw iterations and then incubated at 95℃ for 10 min. The remaining DNA extraction steps were in accordance with the manufacturer's instructions. The extracted DNAs were used as templates in the primary PCR reaction.
The primers used in this study were designed based on the sequence of the SSU rDNA of E. histolytica. For the primary PCR reaction, the primer pairs were used that yielded an expected final product (Table 1). PCR reactions were conducted in a total of 25 µl, which included PCR premix (GenDEPOT, Barker, Texas, USA), 2 µl (~100 ng) of extracted DNA, and 10 pmol of each primer. The PCR reactions were conducted with a G-STORM apparatus (Gene Technologies, Braintree, UK). The DNAs were denatured for 7 min at 94℃, followed by 35 cycles of 60 sec at 94℃, 60 sec at 60℃ or 52℃, and 90 sec at 72℃. Occasionally, the gradient PCR method was used to establish an optimum annealing temperature. Finally, the reactions were incubated for 10 min at 72℃. The nested PCR was conducted with 1 µl of the primary PCR product as the template. After the nested PCR, the final product was electrophoresed in a 2% agarose gel and stained with ethidium bromide. Bands were visualized under ultraviolet light, and photos were taken using a gel documentation system (Syngene, Cambridge, UK).

Primer sets used for detection of Entamoeba histolytica cysts
As shown in Fig. 1A, the primary PCR reaction, using the 1,147 forward and 1,147 reverse primer set, did not show any bands specifically indicating amplified SSU rDNA gene from pure E. histolytica cysts with a final product of 1,147 bp. There was no apparent amplification of the SSU rDNA gene when E. histolytica cysts were added to PBS or stool samples. However, Fig. 1B shows that the nested PCR method specifically amplified 246 bp SSU rDNA in the PBS and also in stools, containing cysts, using the 1,147-246 bp primer set. The nested PCR was conducted using the primers 1,147-246 forward and 1,147-246 reverse set, with the product of the primary PCR products as the template. The detection limit of the nested PCR method for specific amplification of the SSU rDNA gene was 10 pure cysts in PBS, and the same detection limit was found for cysts in stool samples, demonstrating a high sensitivity of this method (Fig. 1B; Table 2). We also conducted nested PCR using a primer pair that specifically amplify the histone H4 gene of E. histolytica; however, no product was amplified from the stool samples (Table 2). Thus, E. histolytica in stool samples could be detected sensitively by amplification of the SSU rDNA gene but not the histone H4 gene.
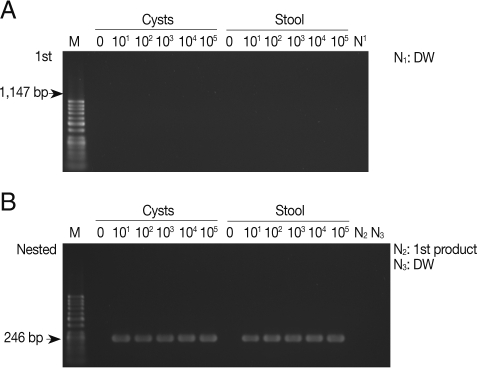
Products of primary and nested PCR amplifications to detect SSU rDNA gene in Entamoeba histolytica cysts. (A) A primary PCR amplification with pure cysts in PBS and in stool samples using the 1,147 primer set. (B) A nested PCR amplification with 1 µl of the primary PCR products as templates using the 1,147-246 primer set. M, DNA marker; N1 and N3, negative control (DW); N2, primary PCR product.

Comparative sensitivity of nested PCR methods using several primer sets
The detection limits of the nested PCR with several primer sets used in this study are shown in Table 2. All nested PCR methods with several primer sets that targeted the SSU rDNA gene had high sensitivities for detecting cysts in PBS. However, most primer sets, with the exception of 1,147-246 bp set, showed useless sensitivity for detection of cysts in stool samples. Only the 1,147-246 primer set showed the highest detection sensitivity for cysts in both PBS and stool samples. Our data strongly suggest that the nested PCR method using the 1,147-246 primer set would be effective for detection of E. histolytica cycsts in clinical samples (Table 2).
The conventional diagnosis of E. histolytica via microscopy has low sensitivity and is prone to give false-positive or false-negative results [12]. Therefore, several PCR-based diagnostic methods have been developed that utilize primers for diverse target genes of E. histolytica [4,6,7,11,13]. The recently developed methods include nested PCR, which is more sensitive than conventional PCR [14,15]. Many different types of factors in clinical samples, such as fecal materials in stool samples, can interfere with PCR analyses, thus, it is needed to use more accurate 2-step nested PCR approaches for clinical diagnosis of amebiasis [16]. The primary PCR may yield only a low concentration of the product, which may lead to an unsuccessful ethidium bromide-based visual detection. However, specific bands can be observed following a secondary PCR using the product of the primary PCR as the template. Our study clearly demonstrated the usefulness of the nested PCR method for detecting E. histolytica SSU rDNA from stool samples.
The Department of Malaria and Parasitic Diseases, Korea National Institute of Health, has undertaken surveys of E. histolytica among patients afflicted with severe diarrhea in 16 city or provincial health institutes and hospitals using an enzyme immunoassay technique. Notably, 0.4-0.5% among 24,000 patients were positive for E. histolytica in 2007 to 2008 [17]. However, the enzyme immunoassay used was suitable only for screening, and more accurate diagnostic methods were necessary. Therefore, we developed a molecular genetic method using the SSU rDNA and histone H4 as candidate genes of E. histolytica. The results strongly suggested that the nested PCR using the 1,147-246 bp primer set to detect SSU rDNA is an effective method for the diagnosis of E. histolytica cysts in stool samples. The present nested PCR method using the 1,147-246 bp primer set revelaed a higher sensitivity than other conventional PCR-based methods, and our method showed specifically amplified DNA bands even with 10 cysts contained in PBS and stool samples [4,6,7]. Moreover, our results indicated that many different types of factors, that may be present in stool samples, do not interfere with the results of our nested PCR method.
In our study, to extract highly pure DNAs of E. histolytica cysts in stool samples, we used an improved version of the formalin-ether concentration technique of Garcia et al. [10,11], and thus could obtain fairly pure DNAs from stool samples. However, a disadvantage of our method is that it is quite complicated and time-consuming. Thus, a simpler and faster DNA extraction method will be necessary.
ACKNOWLEDGEMENTS
We express our sincere gratitudes to Dr. Myeong-Heon Shin of Yonsei University, Korea who kindly provided us with the E. histolytica HM-1 (IMSS c16 strain). This work was supported by a grant from the National Institute of Health (NIH-091-4800-4845-300), National Research and Development Program, Ministry of Health and Welfare, the Republic of Korea.