Abstract
Resistance of Plasmodium spp. to anti-malarial drugs is the primary obstacle in the fight against malaria, and molecular markers for the drug resistance have been applied as an adjunct in the surveillance of the resistance. In this study, we investigated the prevalence of mutations in pvmdr1, pvcrt-o, pvdhfr, and pvdhps genes in temperate-zone P. vivax parasites from central China. A total of 26 isolates were selected, including 8 which were previously shown to have a lower susceptibility to chloroquine in vitro. For pvmdr1, pvcrt-o, and pvdhps genes, no resistance-conferring mutations were discovered. However, a highly prevalent (69.2%), single-point mutation (S117N) was found in pvdhfr gene. In addition, tandem repeat polymorphisms existed in pvdhfr and pvdhps genes, which warranted further studies in relation to the parasite resistance to antifolate drugs. The study further suggests that P. vivax populations in central China may still be relatively susceptible to chloroquine and sulfadoxine-pyrimethamine.
-
Key words: Plasmodium vivax, drug-resistance, mutation, central China
Among the 5 species of
Plasmodium parasites that affect humans,
Plasmodium vivax is the most widely distributed and causes a serious public health burden [
1,
2]. In central China, the transmission of malaria is entirely due to
P. vivax. Since 2000, vivax malaria has demonstrated resilience to eradication and has become increasingly prevalent in this region, especially in areas along the Huai River [
3]. Control measures of vivax malaria are confounded by 2 major factors; dormant hypnozoite stages in the liver, and the emergence of drug resistance. Chemotherapy remains a key factor in the fight against malaria. The tools that are used to monitor drug efficacy include clinical trials, in vitro studies, and molecular markers for detection, each of which made important contributions to a more complete understanding of anti-malarial drug resistance. In a previous study, the susceptibility of
P. vivax to anti-malarial drugs was observed by in vitro testing [
4], though further investigations are needed to assess the predictive value of molecular markers in central China.
Molecular markers of drug-resistant
P. vivax, including mutations in multidrug resistance 1 gene (
pvmdr1) and putative transporter protein CG10 gene (
pvcg10 or
pvcrt-o), which are orthologous to
pfmdr1 and
pfcrt-o genes, respectively, have been identified as possible genetic markers of chloroquine-resistance (CQR) [
5,
6]. The genotypes of dihydrofolate reductase (
pvdhfr) and dihydropteroate synthase (
pvdhps) have been determined, and their association with the clinical response to sulfadoxine-pyrimethamine (SP) has been confirmed [
7-
10]. The sequences which result in amino acid changes of these genes vary depending on the geographic area, and an increasing number of specific mutations results in higher levels of resistance. Chloroquine resistance is associated with single nucleotide polymorphisms (SNPs) in
pvmdr1 at codon (c) 976 (Y976F) [
11] and
pvcrt-o at c10 (K10 insertion). Also, SNPs at c57 (F57L), c58 (S58R), c61 (T61M), and c117 (S117N) in
pvdhfr are associated with pyrimethamine resistance [
12,
13], while sulfadoxine resistance is associated with SNPs at c382 (S382A/C), c383 (383G), and c553 (A553G) [
12]. This study aimed to fill some of the gaps in our knowledge of susceptibility to commonly used anti-malarials, and hence, the prevalence of genetic polymorphisms in
P. vivax drug resistance-associated genes, including
pvmdr1,
pvcrt-o,
pvdhfr, and
pvdhps, were evaluated using isolates from central China.
Based on the completed wild-type sequences, including nucleotides, the amino acid sequences and SNPs reported in the target genes were analyzed; GenBank accession nos. AY571984 for
pvmdr1 [
14], AF314649 for
pvcrt-o [
15], X98123 for
pvdhfr [
8], and AY186730 for
pvdhps [
10]. According to the reported SNP results, the target sequences of each gene were selected for PCR amplification and sequencing, covering the resistance-conferring mutations of each gene. Samples, determined to be positive for
P. vivax by microscopy, were collected from local hospitals and Centers for Disease Control and Prevention (CDC) in central China from 2005 to 2008. Ethical approval was obtained from the ethical review committees at the National Institute of Parasitic Diseases, Chinese CDC, and the Walter Reed Army Institute of Research, USA. Filter papers with whole blood deposits were used for DNA elution by methods described previously [
16]. To avoid cross-contamination, punch was cleaned in 70% ethanol and then punched a clean filter paper 3 times before cutting a new sample. During the process, measures were taken to prevent cross-contamination.
Amplifications of target gene fragments were performed by PCR using 2 µl of 10×PCR buffer (10 mM Tris-HCl, 50 mM KCl, pH 8.3), 2.5 mM MgCl
2, 0.2 mM of each dNTP, 0.25 µM of each primer, 0.5 U of AmpliTaq Gold DNA polymerase (Applied Biosystems, Foster City, California, USA), and 1 µl of genomic DNA or the amplicon from the first PCR. Amplifications were carried out in a G-Storm GSI thermal cycler (Gene Technologies Ltd, Somerton, England) in a final reaction volume of 20 µl. The cycle conditions were an initial denaturation at 94℃ for 10 min, followed by 35 cycles of denaturation at 94℃ for 50 sec, annealing at primer dependent temperature for 1 min, and extension at 72℃ for 1-1.5 min. The gene-specific primers for amplifications were used as described previously (
Table 1). Among them, primers set F1/R1 were used for the first-round PCR to amplify the
pvdhps gene, and F2/R2 were used for the second-round PCR. The purified PCR products were sequenced with the sequencing primer using the ABI PRISM 310 Genetic Analyzer and a Big Dye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). The deduced amino acid sequences were aligned and analyzed with Lasergene® software (DNASTAR, Madison, Wisconsin, USA). Amino acid sequences were compared with the reference wild-type sequences, where insertions and deletions were verified manually.
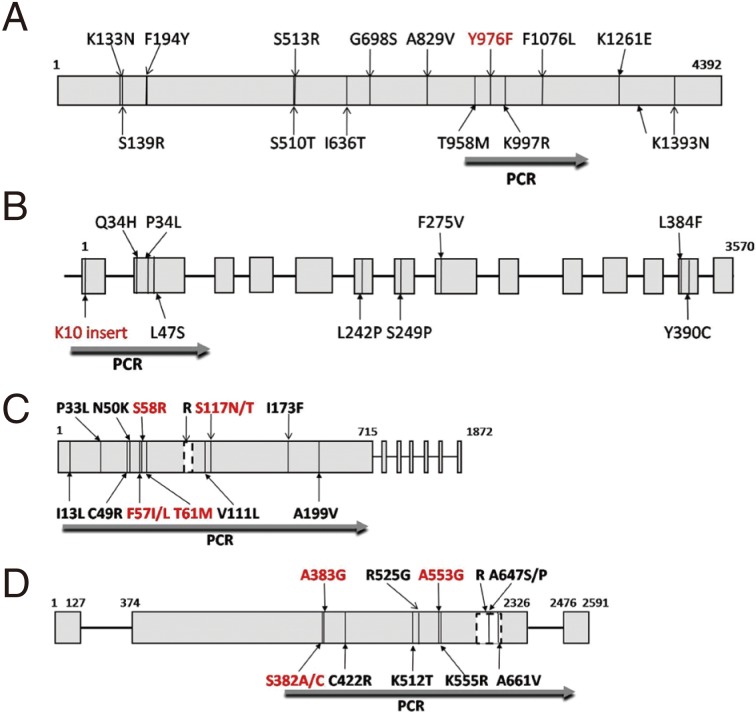
The
pvmdr1 has an open-reading frame (ORF) of 4,392 base pairs (bp) encoding a protein of 1,464 amino acids (aa).
Pvcrt-o coding sequence extends across 14 exons, which range from 45 to 266 bp, each of the exon contains part of the ORF. The whole
pvdhfr-ts gene was composed of an exon with 1,872 bp (623 aa) and a short tandem repeat region (262-309 bp). The DHFR and TS domains of the gene with a linkage sequence of 100 aa encode 237 and 286 aa residues, respectively. The
pvdhps gene consists of 2,591 bp, including the ORF sequence 2,196 bp (731 aa) which includes 3 exons, nucleotides 1-127, 374-2326, and 2,476-2,591 bp, respectively. The reported codon mutations of each gene were indicated, and resistance-conferring mutations were marked with red color (
Fig. 1).
Based on the reported results, fragments of
pvmdr1 (expected amplicon sizes are 604 bp),
pvcrt-o (1,186 bp),
pvdhfr (716 bp), and
pvdhps (1,301 bp) were selected for amplification (
Fig. 1). A total of 26
P. vivax single infection samples were selected for this study, including 8 samples collected in Bengbu, Anhui Province during 2005-2006, which had shown lower susceptibility to chloroquine in a previous in vitro study [
4], as well as 18 further samples collected in Suining, Jiangsu Province during 2008. The history of anti-malarial drugs used in these 2 sites was similar, and Suining is located in the border of the 2 provinces. The
pvmdr1 gene was successfully sequenced in all 26 isolates.
Compared with Salvador I as the reference wild-type, mutant alleles at position 1076 (F1076L) were present in all isolates, wild-type c976 (Y976) was observed in all isolates, and no other non-synonymous or synonymous mutations were found (
Table 2). The
pvcrt-o gene was successfully sequenced in 84.6% (22/26) of the selected isolates, including 8 samples collected in 2005-2006 and 15 samples collected in 2008. Of the target fragment of the
pvcrt-o gene that was sequenced, only wild-type was observed, and the K10 insertion (AAG) did not exist in any of the isolates tested (
Table 2). As
pvmdr1, the
pvdhfr gene was also successfully sequenced and analyzed from all 26 selected isolates. Compared with the wild-type sequence, non-synonymous mutations were detected only at residues 99 and 117. The point mutation at c99 was localized in the short tandem repeat region. The central tandem repeat region between amino acid positions 88 and 105 of the
pvdhfr gene also exhibited size polymorphism in a number of 18 bp repeats.
A total of 23.1% of the samples showed a pattern in the repeat region similar to that in the wild-type. The remaining samples identified as having deletion mutations and previously reported H99S mutations accounted for 65.4% and 11.5%, respectively. The deletion genotype within the
pvdhfr tandem repeat region had the deletion at position 98-103 (THGGDN). The
pvdhfr mutant allele at c117 (S117N) was observed in 18 isolates (69.2%) (
Table 2). The
pvdhps gene was successfully amplified and sequenced from 23 parasite isolates, including 6 samples collected in 2005-2006 and 17 samples collected in 2008. In contrast to the
pvdhfr gene, no SNP was present in
pvdhps except for the tandem repeat variation (spanning 603-665 aa). Length polymorphism of the sequence was observed for a variable number of tandem repeat unit G(E/D)(A/G/S)KLTN, and the samples were identified as 2 kinds of mutation types. The 2 repeat haplotypes were reported previously [
17]. The most common haplotype represented 95.7% (22/23) of the analyzed samples and appeared to be different from the reference wild-type strain by only 1 mutational step. Another haplotype was only present in 1 sample, which was different to the wild-type in the tandem repeat unit and number (
Table 2). The sample with the rare type of tandem repeat in the
dhps gene was identified as having the wild-type
dhfr tandem repeat region. No relationship was observed between the mutations in the sequenced genes and the period of isolate collection.
As the most frequent and widely distributed cause of recurring malaria, P. vivax infection represents a considerable part of malarial disease as well as an economic burden in China, especially in the central region. In China, the action plan for malaria elimination was initiated by the Chinese government in 2010; however, the predominant malaria parasite P. vivax is among the biggest challenges facing the elimination programs. Several possible factors co-contributed to the reemergence of the disease, including emergence of drug resistance and global warming, and hence, it is important to increase research efforts for understanding the epidemiology of P. vivax in this area.
According to the anti-malarial drug policy of China, the first-line therapies for vivax malaria treatment are currently chloroquine plus primaquine during the past 60 years. Clinical resistance to pyrimethamine, used for malaria prophylaxis, was first reported in temperate-zone provinces in the late 1970s [
18,
19], though recent studies suggest that this family of drugs may find a role in the future treatment of
P. vivax malaria in certain regions [
20,
21]. With the emergence of CQR
P. vivax strains in many malarious regions, including the report of treatment failure by chloroquine in 4 cases of
P. vivax malaria in Yunnan Province [
22], it became a much higher priority to monitor drug resistance and develop new drugs for future treatment of
P. vivax malaria. However, there has been no conclusive evidence showing the emergence and spread of CQR
P. vivax in central China until now.
In a previous study, we investigated anti-malarial susceptibility of
P. vivax in this temperate-zone by in vitro testing [
4], and a variety of anti-malarial drugs with potential clinical use in China were tested. The results indicated that for
P. vivax isolates from this area, the IC
50s of chloroquine were lower than those of isolates from South Korea and Thailand. Reduced susceptibility to pyrimethamine was also observed, compared to isolates from Thailand, but a greater sensitivity was observed in isolates from South Korea. Four patients from the same area were enrolled in the study, each of which had parasites with higher in vitro IC
50s, suggesting the presence of clinical resistance to chloroquine in this area. The molecular data for
pvmdr1,
pvcrt-o,
pvdhfr, and
pvdhps in this study could provide useful information about drug resistance in
P. vivax isolates from central China.
Although the mechanisms of CQR in
P. vivax are not very clear, 2 main transporters have been studied with regard to CQR in
P. vivax, including
pvcrt-o and
pvmdr1, indicating that the amino acid polymorphisms were associated with chloroquine resistance [
9,
10]. In our study,
pvmdr1 Y976F mutation and K10 insertion in the
pvcrt-o gene were not found in all of the isolates tested, including those which had shown higher chloroquine IC
50 values through an in vitro drug sensitivity study. Thus, it is indicated that CQR may not yet be prevalent in central China as had been previously thought. The point mutation
pvmdr1 F1076L, which has been suggested to be a neutral allele [
23], existed in all isolates tested.
The resistance mechanism of
P. vivax to antifolates has been proposed to be similar to that of
P. falciparum, which has been linked to mutations at homologous positions in
pvdhfr and
pvdhps [
11-
14]. Results from in vitro drug assays and clinical assessments are generally agreeable with this assumption [
12,
24-
27], suggesting that molecular genotyping data for
pvdhfr and
pvdhps could provide useful information about SP resistance in
P. vivax. Our study showed that mutant
pvdhfr genotypes were present at relatively high levels, but the resistance-conferring mutations only occurred at a single c117N. Two mutant genotypes at residue 117 (S117N and S117T) have been reported in areas with extensive SP drug treatment [
10,
28] and only S117N was observed in this study. The S117N mutation has been hypothesized to represent the first step in the drug-resistance selection process in the parasite [
29]. In contrast, S117T has been associated with highly mutated
pvdhfr, which may be a key mutation for subsequent acquisition of additional mutations and development of high resistance to SP [
11,
29]. Consistent with these, highly mutated
pvdhfr (double, triple, or quadruple mutations) was not observed in this study. Similar to a previous report, no resistance-conferring mutations were found in
pvdhps gene among all of the isolates examined in this study. It has been proposed that the development of resistance mutations in
dhfr and
dhps of
P. vivax is asynchronous, and mutations in
dhfr appear to be selected before those in
dhps [
25,
30]. The present study showed that resistance-conferring mutations were found in
pvdhfr but not in
pvdhps and thus further validated this hypothesis. The results suggest that the
P. vivax parasites in central China may be still relatively susceptibility to SP.
A tandem repeat sequence is one of the unique features in the
pvdhfr, which is absent in
P. falciparum and
P. chabaudi. The isolates examined in this study were typically separated into 2 types, deletion (nucleotides 292-309) and mutation H99S in
pvdhfr, with the exception of 6 wild-type isolates. Both types of the tandem repetitive sequence did not appear to be clearly associated with a specific genetic profile, and no relationship was found between the type of c117 and the repeat. A similar repeat motif has also been described in
pvdhps gene [
31,
32], which has been predicted not to directly bind sulfa drugs, and therefore is unlikely to be involved in resistance to sulfa drugs in
P. vivax. No wild-type of the tandem repeat sequence was determined in the
pvdhps gene in this study. They were typically separated in 2 types also, although 1 type (95.7%) was mainly observed. Both of these tandem repeat sequences showed size polymorphisms, which may indicate a lack of SP-selective sweep in these parasite populations, and warrants further studies of the parasite resistance to antifolate drugs.
Resistance-conferring mutations in
pvmdr1,
pvcrt-o, and
pvdhps genes were not discovered, but a single point mutation in
pvdhfr gene was common to both studies. Comparing the results with isolates from South-East Asia, temperate
P. vivax strains show much lower polymorphisms in these drug-resistance genes, consistent with the drugs pressure in these areas [
28,
33]. Although the number of samples used in this study was limited, isolates which had shown a likelihood of drugs resistance were selected, and the results were shown to be comparable to other studies in central China [
17,
25,
34]. Analysis of the SNPs of drug-resistance genes has been proven to be useful and important in monitoring drug resistance in malaria endemic countries and should be investigated thoroughly.
ACKNOWLEDGMENTS
This work was supported by National Research Foundation of Korea Grant funded by the Korean Government (2009-0075103) and Mid-Career Researcher Program through NRF grand funded by the MEST (2011-0016401) and grants from the National Basic Research Programme (973 Programme) in China (2007CB513100), the China National S & T Major Programme (Grant No. 2008ZX10004-011).
References
- 1. Anstey NM, Russell B, Yeo TW, Price RN. The pathophysiology of vivax malaria. Trends Parasitol 2009;25:220-227.
- 2. Mueller I, Galinski MR, Baird JK, Carlton JM, Kochar DK, Alonso PL, del Portillo HA. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect Dis 2009;9:555-566.
- 3. Zhou SS, Wang Y, Tang LH. Malaria situation in the People's Republic of China in 2006. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi 2007;25:439-441. (in Chinese).
- 4. Lu F, Gao Q, Chotivanich K, Xia H, Cao J, Udomsangpetch R, Cui L, Sattabongkot J. In vitro anti-malarial drug susceptibility of temperate Plasmodium vivax from central China. Am J Trop Med Hyg 2011;85:197-201.
- 5. Suwanarusk R, Russell B, Chavchich M, Chalfein F, Kenangalem E, Kosaisavee V, Prasetyorini B, Piera KA, Barends M, Brockman A, Lek-Uthai U, Anstey NM, Tjitra E, Nosten F, Cheng Q, Price RN. Chloroquine resistant Plasmodium vivax: In vitro characterisation and association with molecular polymorphisms. PLoS One 2007;2:e1089.
- 6. Brega S, Meslin B, de Monbrison F, Severini C, Gradoni L, Udomsangpetch R, Sutanto I, Peyron F, Picot S. Identification of the Plasmodium vivax mdr-like gene (pvmdr1) and analysis of single-nucleotide polymorphisms among isolates from different areas of endemicity. J Infect Dis 2005;191:272-277.
- 7. Tjitra E, Baker J, Suprianto S, Cheng Q, Anstey NM. Therapeutic efficacies of artesunate-sulfadoxine-pyrimethamine and chloroquine-sulfadoxine-pyrimethamine in vivax malaria pilot studies: Relationship to Plasmodium vivax dhfr mutations. Antimicrob Agents Chemother 2002;46:3947-3953.
- 8. de Pécoulas PE, Tahar R, Ouatas T, Mazabraud A, Basco LK. Sequence variations in the Plasmodium vivax dihydrofolate reductase-thymidylate synthase gene and their relationship with pyrimethamine resistance. Mol Biochem Parasitol 1998;92:265-273.
- 9. Imwong M, Pukrittakayamee S, Looareesuwan S, Pasvol G, Poirreiz J, White NJ, Snounou G. Association of genetic mutations in Plasmodium vivax dhfr with resistance to sulfadoxine-pyrimethamine: geographical and clinical correlates. Antimicrob Agents Chemother 2001;45:3122-3127.
- 10. Korsinczky M, Fischer K, Chen N, Baker J, Rieckmann K, Cheng Q. Sulfadoxine resistance in Plasmodium vivax is associated with a specific amino acid in dihydropteroate synthase at the putative sulfadoxine-binding site. Antimicrob Agents Chemother 2004;48:2214-2222.
- 11. Phillips EJ, Keystone JS, Kain KC. Failure of combined chloroquine and high-dose primaquine therapy for Plasmodium vivax malaria acquired in Guyana, South America. Clin Infect Dis 1996;23:1171-1173.
- 12. Imwong M, Pukrittayakamee S, Cheng Q, Moore C, Looareesuwan S, Snounou G, White NJ, Day NP. Limited polymorphism in the dihydropteroate synthetase gene (dhps) of Plasmodium vivax isolates from Thailand. Antimicrob Agents Chemother 2005;49:4393-4395.
- 13. Marfurt J, de Monbrison F, Brega S, Barbollat L, Müller I, Sie A, Goroti M, Reeder JC, Beck HP, Picot S, Genton B. Molecular markers of in vivo Plasmodium vivax resistance to amodiaquine plus sulfadoxine-pyrimethamine: mutations in pvdhfr and pvmdr1. J Infect Dis 2008;198:409-417.
- 14. Sá JM, Nomura T, Neves J, Baird JK, Wellems TE, del Portillo HA. Plasmodium vivax: allele variants of the mdr1 gene do not associate with chloroquine resistance among isolates from Brazil, Papua, and monkey-adapted strains. Exp Parasitol 2005;109:256-259.
- 15. Nomura T, Carlton JM, Baird JK, del Portillo HA, Fryauff DJ, Rathore D, Fidock DA, Su X, Collins WE, McCutchan TF, Wootton JC, Wellems TE. Evidence for different mechanisms of chloroquine resistance in 2 Plasmodium species that cause human malaria. J Infect Dis 2001;183:1653-1661.
- 16. Lu F, Gao Q, Zhou H, Cao J, Wang W, Lim CS, Na S, Tsuboi T, Han ET. Molecular test for vivax malaria with loop-mediated isothermal amplification method in central China. Parasitol Res 2012;110:2439-2444.
- 17. Miao M, Yang Z, Cui L, Ahlum J, Huang Y. Different allele prevalence in the dihydrofolate reductase and dihydropteroate synthase genes in Plasmodium vivax populations from China. Am J Trop Med Hyg 2010;83:1206-1211.
- 18. Chen JS, Zhang KR, Geng ZW. Clinical observation on the in vivo sensitivity of Plasmodium vivax to pyrimethamine in Kaifeng District, Henan Province. Henan J Prev Med 1977;51-56. (in Chinese).
- 19. Shen JQ, Hu XS. Comparison of efficacy between long-term oral administration of pyrimethamine and antimalarial pill 3. Railway Med J 1978;2:90-91. (in Chinese).
- 20. Hastings MD, Sibley CH. Pyrimethamine and WR99210 exert opposing selection on dihydrofolate reductase from Plasmodium vivax. Proc Natl Acad Sci USA 2002;99:13137-13141.
- 21. Hawkins VN, Joshi H, Rungsihirunrat K, Na-Bangchang K, Sibley CH. Antifolates can have a role in the treatment of Plasmodium vivax. Trends Parasitol 2007;23:213-222.
- 22. Yang XM, Yang MQ, Huang JW. Clinical research of the sensitivity of Plasmodium vivax to chloroquine. Chinese J Parasitol Parasit Dis 1996;9:226-227. (in Chinese).
- 23. Imwong M, Pukrittayakamee S, Pongtavornpinyo W, Nakeesathit S, Nair S, Newton P, Nosten F, Anderson TJ, Dondorp A, Day NP, White NJ. Gene amplification of the multidrug resistance 1 gene of Plasmodium vivax isolates from Thailand, Laos, and Myanmar. Antimicrob Agents Chemother 2008;52:2657-2659.
- 24. Hastings MD, Maguire JD, Bangs MJ, Zimmerman PA, Reeder JC, Baird JK, Sibley CH. Novel Plasmodium vivax dhfr alleles from the Indonesian Archipelago and Papua New Guinea: association with pyrimethamine resistance determined by a Saccharomyces cerevisiae expression system. Antimicrob Agents Chemother 2005;49:733-740.
- 25. Auliff A, Wilson DW, Russell B, Gao Q, Chen N, Anh le N, Maguire J, Bell D, O'Neil MT, Cheng Q. Amino acid mutations in Plasmodium vivax DHFR and DHPS from several geographical regions and susceptibility to antifolate drugs. Am J Trop Med Hyg 2006;75:617-621.
- 26. Rungsihirunrat K, Na-Bangchang K, Hawkins VN, Mungthin M, Sibley CH. Sensitivity to antifolates and genetic analysis of Plasmodium vivax isolates from Thailand. Am J Trop Med Hyg 2007;76:1057-1065.
- 27. Hastings MD, Porter KM, Maguire JD, Susanti I, Kania W, Bangs MJ, Sibley CH, Baird JK. Dihydrofolate reductase mutations in Plasmodium vivax from Indonesia and therapeutic response to sulfadoxine plus pyrimethamine. J Infect Dis 2004;189:744-750.
- 28. Lu F, Lim CS, Nam DH, Kim K, Lin K, Kim TS, Lee HW, Chen JH, Wang Y, Sattabongkot J, Han ET. Mutations in the antifolate-resistance-associated genes dihydrofolate reductase and dihydropteroate synthase in Plasmodium vivax isolates from malaria-endemic countries. Am J Trop Med Hyg 2010;83:474-479.
- 29. Brega S, de Monbrison F, Severini C, Udomsangpetch R, Sutanto I, Ruckert P, Peyron F, Picot S. Real-time PCR for dihydrofolate reductase gene single-nucleotide polymorphisms in Plasmodium vivax isolates. Antimicrob Agents Chemother 2004;48:2581-2587.
- 30. Sibley CH, Hyde JE, Sims PF, Plowe CV, Kublin JG, Mberu EK, Cowman AF, Winstanley PA, Watkins WM, Nzila AM. Pyrimethamine-sulfadoxine resistance in Plasmodium falciparum: what next? Trends Parasitol 2001;17:582-588.
- 31. Menegon M, Majori G, Severini C. Genetic variations of the Plasmodium vivax dihydropteroate synthase gene. Acta Trop 2006;98:196-199.
- 32. Hawkins VN, Suzuki SM, Rungsihirunrat K, Hapuarachchi HC, Maestre A, Na-Bangchang K, Sibley CH. Assessment of the origins and spread of putative resistance-conferring mutations in Plasmodium vivax dihydropteroate synthase. Am J Trop Med Hyg 2009;81:348-355.
- 33. Lu F, Lim CS, Nam DH, Kim K, Lin K, Kim TS, Lee HW, Chen JH, Wang Y, Sattabongkot J, Han ET. Genetic polymorphism in pvmdr1 and pvcrt-o genes in relation to in vitro drug susceptibility of Plasmodium vivax isolates from malaria-endemic countries. Acta Trop 2011;117:69-75.
- 34. Huang F, Zhou S, Zhang S, Li W, Zhang H. Monitoring resistance of Plasmodium vivax: point mutations in dihydrofolate reductase gene in isolates from Central China. Parasit Vectors 2011;4:80.
Fig. 1Schematic diagrams of amino acid point mutation positions and sequencing regions of pvmdr1 (A), pvcrt-o (B), pvdhfr (C), and pvdhps (D) genes. Previously reported point mutations are indicated, and resistance-conferring mutations are marked in red. B and D; boxes indicate exons, and lines indicate introns. R; repeat region GGDN/TSGGDN/THGGDN (C) and GEAKLTN-GEGKLTN-GEAKLTN-GEGKLTN-GEAKLTN-GEGKLTN-GDAKLTN-GDSKLTN-GEAKLTN (D).
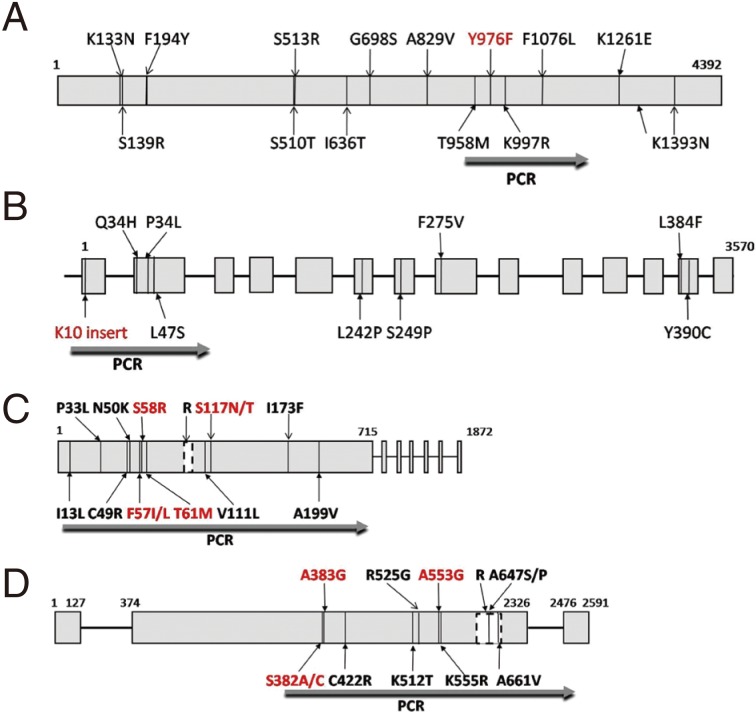
Table 1.Sequence and features of primers used in this study
Table 1.
|
Gene |
Primer sequence (5´→3´) |
Position (bp)a
|
Use |
Amplicon size (bp) |
Annealing temp. (°C) |
Reference |
|
Pvmdr1
|
F: GGATAGTCATGCCCCAGGATTG |
2751--2772 |
PCR/Seq |
604 |
62 |
[9] |
|
R: CATCAACTTCCCGGCGTAGC |
3335--3354 |
PCR |
|
|
|
|
Pvcrt-o
|
F: AAGAGCCGTCTAGCCATCC |
−99--−81 |
PCR/Seq |
1,186 |
61 |
[35] |
|
R: AGTTTCCCTCTACACCCG |
1069--1086 |
PCR |
|
|
|
|
Pvdhfr
|
F: ATGGAGGACCTTTCAGATGTATT |
1--23 |
PCR/Seq |
716 |
58 |
[11] |
|
R:CCACCTTGCTGTAAACCAAAAAGTCCAGAG |
686--715 |
PCR |
|
|
|
|
Pvdhps
|
F1:AGGAAGCCATTCGCTCAAC |
1121--1139 |
PCR |
1,700 |
56 |
[34] |
|
R1:GGAACGCTGCAAACAACAC |
+211--+229 |
PCR |
|
|
|
|
F2:GGTTTATTTGTCGATCCTGTG |
1300--1320 |
PCR/Seq |
1,301 |
56 |
[14] |
|
R2:GAGATTACCCTAAGGTTGATGTATC |
2576--+9 |
PCR/Seq |
|
|
|
Table 2.Prevalence of single nucleotide polymorphisms and tandem repeat genotypes in pvmdr1, pvcrt-o, pvdhfr, and pvdhps genes in central China
Table 2.
|
Genotype in each drug resistant gene |
No. of isolates sequenced/no. of total isolate selected (%) |
|
Pvmdr1
|
|
|
Wild-type Y976 codon |
26/26 (100) |
|
Mutant L1076 codon |
26/26 (100) |
|
Pvcrt-o
|
|
|
Wild-type without K10 insert |
22/22 (100) |
|
Pvdhfr
|
|
|
Wild-type genotype (IPCNFSTVSIA) |
8/26 (30.8) |
|
Mutant N117 codon |
18/26 (69.2) |
|
Mutant tandem repeata
|
20/26 (76.9) |
|
Pvdhps
|
|
|
Wild-type genotype (SACKAVA) |
23/23 (100) |
|
Mutant tandem repeatb
|
23/23 (100) |