Genetic Characterization of Clinical Acanthamoeba Isolates from Japan using Nuclear and Mitochondrial Small Subunit Ribosomal RNA
Article information
Abstract
Because of an increased number of Acanthamoeba keratitis (AK) along with associated disease burdens, medical professionals have become more aware of this pathogen in recent years. In this study, by analyzing both the nuclear 18S small subunit ribosomal RNA (18S rRNA) and mitochondrial 16S rRNA gene loci, 27 clinical Acanthamoeba strains that caused AK in Japan were classified into 3 genotypes, T3 (3 strains), T4 (23 strains), and T5 (one strain). Most haplotypes were identical to the reference haplotypes reported from all over the world, and thus no specificity of the haplotype distribution in Japan was found. The T4 sub-genotype analysis using the 16S rRNA gene locus also revealed a clear sub-conformation within the T4 cluster, and lead to the recognition of a new sub-genotype T4i, in addition to the previously reported sub-genotypes T4a-T4h. Furthermore, 9 out of 23 strains in the T4 genotype were identified to a specific haplotype (AF479533), which seems to be a causal haplotype of AK. While heterozygous nuclear haplotypes were observed from 2 strains, the mitochondrial haplotypes were homozygous as T4 genotype in the both strains, and suggested a possibility of nuclear hybridization (mating reproduction) between different strains in Acanthamoeba. The nuclear 18S rRNA gene and mitochondrial 16S rRNA gene loci of Acanthamoeba spp. possess different unique characteristics usable for the genotyping analyses, and those specific features could contribute to the establishment of molecular taxonomy for the species complex of Acanthamoeba.
INTRODUCTION
The genus Acanthamoeba has been isolated from various environmental samples such as soil [1], water [2], air [3], and also human nasal mucosa [4]. While, during the last few decades, this ubiquitous free-living amoeba [5] has become increasingly recognized as a causal agent of serious human diseases, such as vision-threatening Acanthamoeba keratitis (AK), life-threatening granulomatous amoebic encephalitis, and disseminated infections of other tissues [6].
Due to an increased number of Acanthamoeba infections along with associated disease burdens, medical professionals have become more aware of this pathogen in recent years [7]. Since 1973 when the first case was reported in a contact lens wearer (CLW), AK has been reported from all over the world [8]. While the prevalence of AK was shown to vary from 1 per 10,000 to 1,000,000 CLW [7,9], the infection clearly appears to be dominant in CLW; on the other hand, the cases of AK in non-CLW are quite limited [10]. To date, the numbers of clinical cases worldwide have been increased as consequently gained the disease recognition [11]. Such a trend has also been observed in Japan, since the first case reported in 1988 [12].
The previous classification of Acanthamoeba spp., especially using morphology, caused various ambiguities and therefore has been revised several times [13]. In the early time, classification trials divided this species into 3 groups (I, II, and III) according to the cyst size and shape [14], which, however, was criticized by later studies showing numerous inconsistencies between the morphological classification and previous species categories [13,15]. The current molecular classification divides Acanthamoeba spp. into 15 haplotypes (T1-T15), based on nucleotide sequence variations in the 18S rRNA gene [16]. While 2 additional genotypes, T16 and T17, have been recently reported [17,18], the number of these isolates is still limited. Therefore, far more reference information is required to confirm these novel clusters.
Among a total of 15 (or 17) genotypes, the majority of clinical and environmental isolates of amoeba belong to the T4 genotype [13,19]; however, phylogenetic reconstructions of the T4 sub-genotypes were problematic, due to low resolutions of the 18S rRNA gene [13]. On the other hand, the mitochondrial 16S rRNA gene locus seems to have some promising characteristics for the T4 sub-genotype analysis [20,21]. In addition, the 16S rRNA gene locus contains no intron and has more diversity than the 18S rRNA gene locus. However, the number of mitochondrial gene references is still quite limited.
In this study, the genetic diversity of Acanthamoeba spp. isolated from keratitis patients was examined using both the nuclear and mitochondrial gene loci. Our results reveal the detailed diversity of T4 sub-genotypes.
MATERIALS AND METHODS
Isolates and culture condition
Twenty-seven cultured Acanthamoeba spp. isolates were used in this study. The samples JPH1 to JPH8 were reference isolates provided from the National Institutes of Infectious Diseases (Tokyo, Japan), originally isolated from AK patients all over Japan. While the samples JPH9 to JPH27 were collected from AK patients between 2006 and 2009, at Kanazawa Medical University and Kanazawa University Hospital, Ishikawa, Japan, and have been culturally maintained in our group. As the culture medium, an amoeba saline containing 0.012% NaCl, 0.00035% KCl, 0.0003% CaCl2, and 0.0004% MgCl2, 7 H2O in 0.05 mM Tris-HCl (pH 6.8) supplemented with inactivated Escherichia coli [22] was used, and maintained at 27℃.
DNA extraction
Cultured samples were centrifuged at 8,000 rpm for 5 min at 4℃. From the pellet fraction, the whole cell DNA was extracted using QuickGene™ DNA tissue kit S (FUJIFILM Corporation, Tokyo, Japan) according to the manufacturer's instructions, and concentrated by an ethanol precipitation method. The DNA was preserved as an aqueous solution at -20℃ until use.
18S rRNA PCR
A partial DNA fragment (537-560 bp) of the nuclear 18S rRNA gene of Acanthamoeba was amplified using a modified primer set (YKF2/JDF2, Table 1) based on previously published primers [19] on MyCycler™ (BioRad Laboratories, Hercules, California, USA). PCR amplifications were carried out in 20 µl reaction mixture as 1×PrimeSTAR buffer containing a 1-2 µl of the extracted Acanthamoeba DNA template solution, 0.8 mM of each deoxynucleoside triphosphate (dNTP), 0.3 µM of primers, and 1 U of PrimeSTAR HS DNA polymerase (Takara Bio Inc., Shiga, Japan). The PCR cycling profile consisted of 98℃ for 2 min, followed by 35 cycles of 98℃ for 10 sec, an annealing temperature of 63℃ for 5 sec, and 72℃ for 40 sec, then a final extension of 72℃ for 5 min. The PCR products were electrophoresed on 1.2% L03 agarose (TaKaRa) with ethidium bromide, and visualized on a UV transilluminator, Gel Doc™ EZ Imager (BioRad Laboratories). The target bands were then excised from the gel and purified using the Quantum Prep™ Freeze 'N Squeeze DNA Gel Extraction Spin Columns (BioRad Laboratories) according to the manufacturer's instructions.

Primers used in this study
16S rRNA PCR
A partial DNA fragment (1,507-1,533 bp) of the mitochondrial 16S rRNA gene of Acanthamoeba was amplified using a primer set with the reference of previously published primers (FP16/RP16, Table 1) [20]. The PCR conditions and following visualization and purification procedures of the amplicons were the same as used for 18S rRNA PCR. The PCR cycling profile consisted of 98℃ for 2 min, followed by 30 cycles of 98℃ for 10 sec, an annealing temperature of 62℃ for 5 sec, and 72℃ for 95 sec, then a final extension of 72℃ for 5 min.
DNA sequencing
Generally, the purified PCR products were directly sequenced with amplification primers and/or appropriate sequencing primers in both directions on Applied Biosystems 3130 Genetic Analyzer by using the ABI Prism BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Tokyo, Japan). While, due to mixed-nucleotide profiles observed in the sequence data of JPH5 and JPH17, a subcloning strategy was adopted to confirm the sequences. Those PCR amplicons were cloned into the EcoRV site of pBluescript II SK(+) (Stratagene, La Jolla, California, USA) using the Mighty Cloning Reagent Set (bluntend) (Takara Bio Inc, Shiga, Japan). The recombinant plasmids were transformed into Escherichia coli DH5α (Stratagene) and screened on Luria Broth (LB) agar plates supplemented with 100 mg/L of ampicillin. The clones were picked up as E. coli DH5α colonies on the plate and cultured overnight in the 2 ml LB supplemented with 100 mg/L of ampicillin. From the E. coli pellet, plasmid purification was conducted using the QIAGEN® Plasmid Mini Kit (QIAGEN K.K., Tokyo, Japan) according to the manufacturer's instructions, and their full-length sequences were confirmed using T3 and T7 primers. All DNA sequences were assembled using the DNASIS-Mac v3.6 (Hitachi, Yokohama, Japan) and confirmed in both directions.
Sequence alignment and phylogenetic analysis
All reference sequences of the 18S rRNA and 16S rRNA genes of Acanthamoeba used in this study were obtained from the DNA Data Bank of Japan (DDBJ) using the blastn algorithm (http://blast.ddbj.nig.ac.jp/top-e.html). DNA sequence alignments were performed by Clustal W2 v2.1 on the European Bioinformatics Institute (EBI) homepage (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The phylogenetic reconstructions of neighbor-joining (NJ), maximum parsimony (MP), and the maximum likelihood (ML) methods were conducted by MEGA5 [23] and used for its comparative analysis. To construct NJ, MP, and ML phylogenetic trees, we used the same options as bootstrap method with 1,000 of replications, nucleotide substitution type, and complete deletion of gaps/missing data treatment. The other and specific options we used were maximum composite likelihood model and uniform rates in NJ analysis, close-neighbor-interchange (CNI) on random trees of MP search method and MP search level 1 in MP analysis; and general time reversible model and nearest-neighbor-interchange (NNI) of ML heuristic method in ML analysis. We also estimated the average evolutionary divergence of partial sequences of 18S rRNA gene (25 sequences including 2 subclone variations) and 16S rRNA gene (23 sequences) within the T4 genotype confirmed in this study by MEGA5 [23] with the options for the distance analysis preference; bootstrap method with 1,000 of replications from variance estimation method, nucleotide substitution type, maximum composite likelihood model, and complete deletion of gaps/missing data treatment. As the result of this option, the final sequence length used in the analysis were approximately 500 bp.
Nucleotide sequence accession numbers
All the newly identified partial sequences of the 18S rRNA and 16S rRNA genes of Acanthamoeba in the present study were deposited in the DDBJ/European Molecular Biology Laboratory (EMBL)/GenBank nucleotide sequence databases under accession numbers of AB741044-AB741047, AB795719, and AB795705-AB795718, respectively.
RESULTS
Microscopic examination
In all 27 cultured Acanthamoeba spp. samples, we observed 2 life cycle stages, an active trophozoite stage and a dormant cyst stage. After inoculation of the cyst forms, it took 2-12 days (average, 3.4 days) to detect trophozoites by microscopic examinations. The trophozoites, approximately 15-40 µm in diameter, were found to produce many spine-like processes, whereas the cysts, approximately 10-20 µm in diameter, typically had wrinkled double walls and were almost round in shape (Fig. 1).
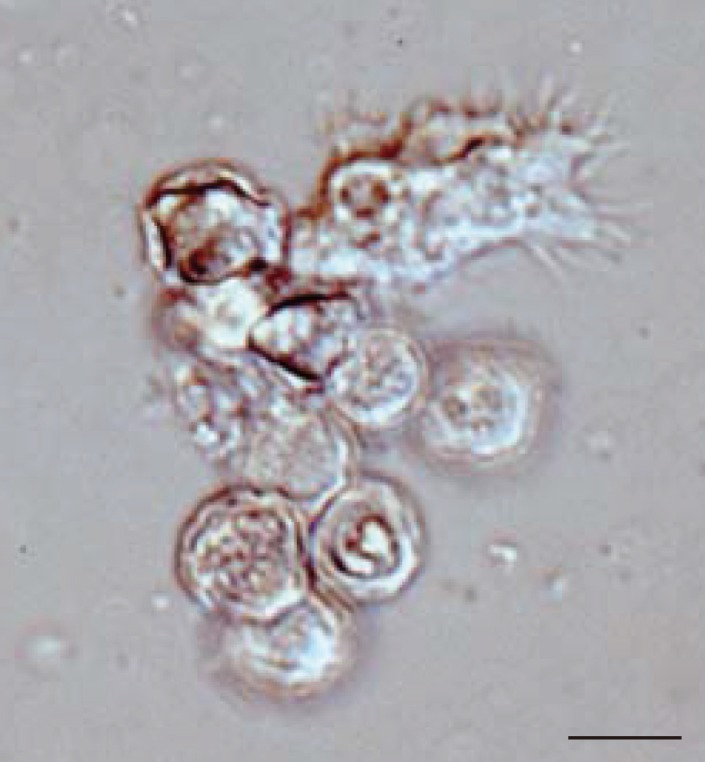
Representative image of culture-isolated Acanthamoeba sp. from an AK case (JPH9). Nomarski interference contrast micrograph of cysts showing double-layered walls and a trophozoite showing spike-like pseudopodia (acanthopodia). Scale bar=10 µm.
18S rRNA gene analysis
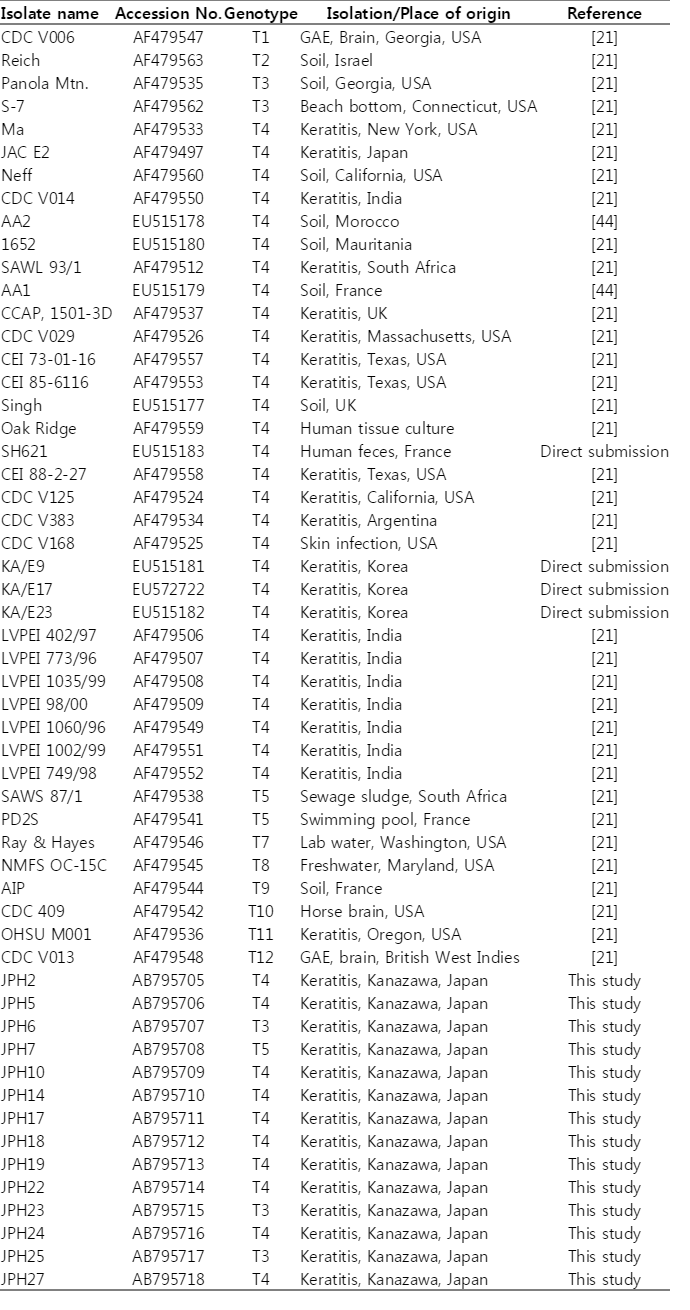
The 18S rRNA gene segments (517-570 bp) were successfully amplified by PCR from all the 27 samples. Of these amplicons, 25 sequences were confirmed by direct sequencing using the amplification primers, whereas the other 2, JPH5 and JPH17, showed sequence heterogeneity. To separate mixed haplotypes from the amplicons of JPH5 and JPH17, we used the subcloning procedure described in "Materials and Methods". The results confirmed the presence of 2 clones in each amplicon, designated as JPH5 (A and B) and JPH17 (A and B). In total, 29 sequences of the 18S rRNA gene from 27 isolates were confirmed. As the results of phylogenetic reconstruction, these 29 sequences and previous reference sequences formed 3 monophyletic clusters, T3, T4, and T5, with significant bootstrap values (100%, 81%, and 100%, respectively, by the NJ method) (Fig. 2). In the T3 cluster, JPH6, 23, and 25 showed 100% identity to the reference sequences S81337, GQ397466, and GQ905499, respectively. In the T4 cluster, several groups also showed 100% identity to their respective reference sequences (accession no. in parenthesis): JPH10, 16, and 21 (U07410); JPH26 (AY703004); JPH3, 12, 15 17B, and 20 (U07403); JPH4 (U07413); JPH27 (AY148954); JPH11 (GU808328); JPH1, 9, and 17A (U07415); JPH5B and 22 (AY173000); JPH2 and 14 (GU936484); and JPH5A and 19 (U07408). As for the T5 cluster, only JPH7 was clustered with 98% homology to the reference sequence of A. lenticulata 25/1 (U94740).
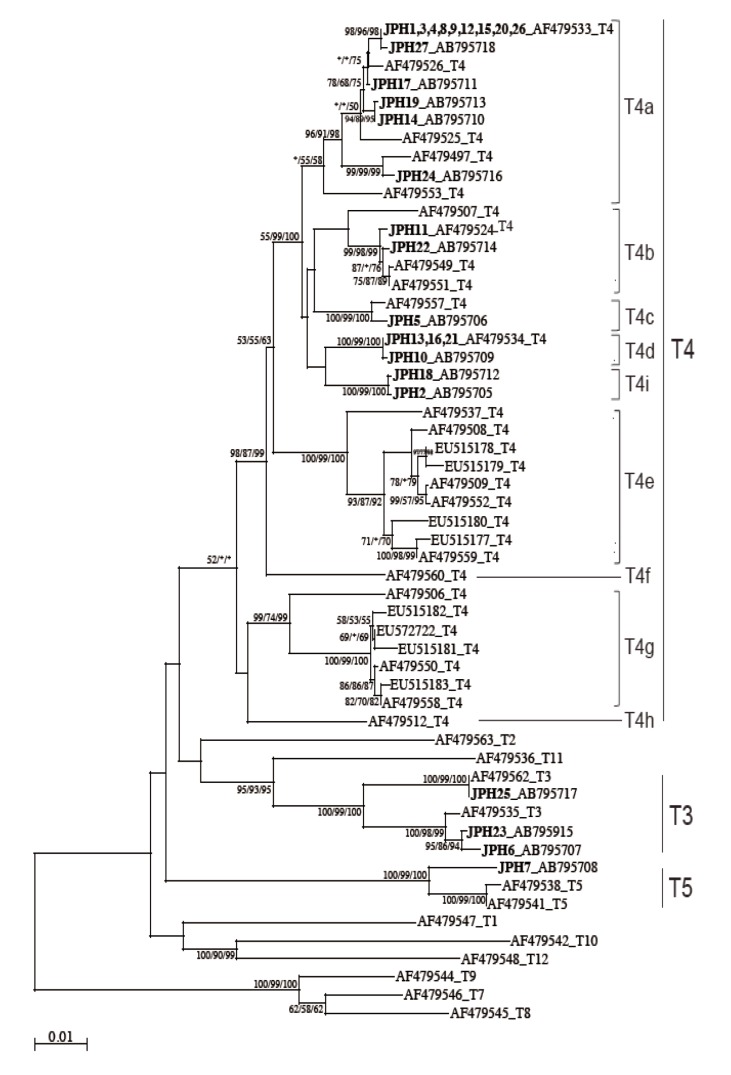
Neighbor-joining (NJ) tree reconstructed with the 18S rDNA sequences of Acanthamoeba. The evolutionary history was inferred using the NJ method as described in "Materials and Methods". Isolates from Acanthamoeba keratitis are shown in boldface text, mixed infections with underlines, and with new or reference accession numbers. All reference sequences are shown with the accession numbers and the genotypes. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap value (1,000 replicates) are shown next to the branches. The evolutionary distances are shown in the units of the number of base substitutions per site.
16S rRNA gene analysis
The 16S rRNA gene segments (1,520-1,545 bp) were also successfully PCR amplified from all the 27 samples, and subsequently, all the sequences were confirmed by direct sequencing. Unlike the 18S rRNA gene analysis results, no sequence heterogeneity was observed in any of the strains analyzed. The reconstructed neighbor-joining tree further showed that these 27 sequences of the 16S rRNA gene and previous reference sequences formed 2 monophyletic clusters, T3 and T5, with significant bootstrap values (100/99/100% and 100/99/100%, respectively, by NJ/MP/ML methods) (Fig. 3). Although the whole T4 cluster was not statistically supported by the bootstrap values (52/<50/<50%), the individual T4 subclusters were significantly supported as follows: T4a (96/91/98%, excluding the reference AF479553), T4b (99/98/99%, excluding the reference AF479507), T4c (100/99/100%), T4d (100/99/100%), T4e (100/99/100%), T4g (99/74/99%), and T4i (100/99/100%). The bootstrap values were not calculated for T4f and T4h, 2 previously proposed subclusters [21], since none of the 27 samples analyzed in this study was clustered with the reference sequences. Based on the phylogenetic reconstruction results, all the 27 sequences were classified as specific genotypes as follows: T3 cluster, JPH6 (AB795707), JPH23 (AB795915), and JPH25 (AB795717); T4a cluster, JPH1, 3, 4, 8, 9, 12, 15, 20, and 26 (AF479533), JPH27 (AB795718), JPH17 (AB795711), JPH19 (AB795713), JPH14 (AB795710), and JPH24 (AB795716); T4b cluster, JPH11 (AF479524) and JPH22 (AB795714); T4c cluster, JPH5 (AB795706); T4d cluster, JPH13, 16, and 21 (AF479534), and JPH10 (AB795709); and T4i cluster, JPH18 (AB795712) and JPH2 (AB795705). In the T5 cluster, only JPH7 was clustered with 97% homology to the reference sequence of A. lenticulata PD2S (AF479541). Overall, there was no dissimilarity between the genotyping results of 18S rRNA and 16S rRNA gene analyses.
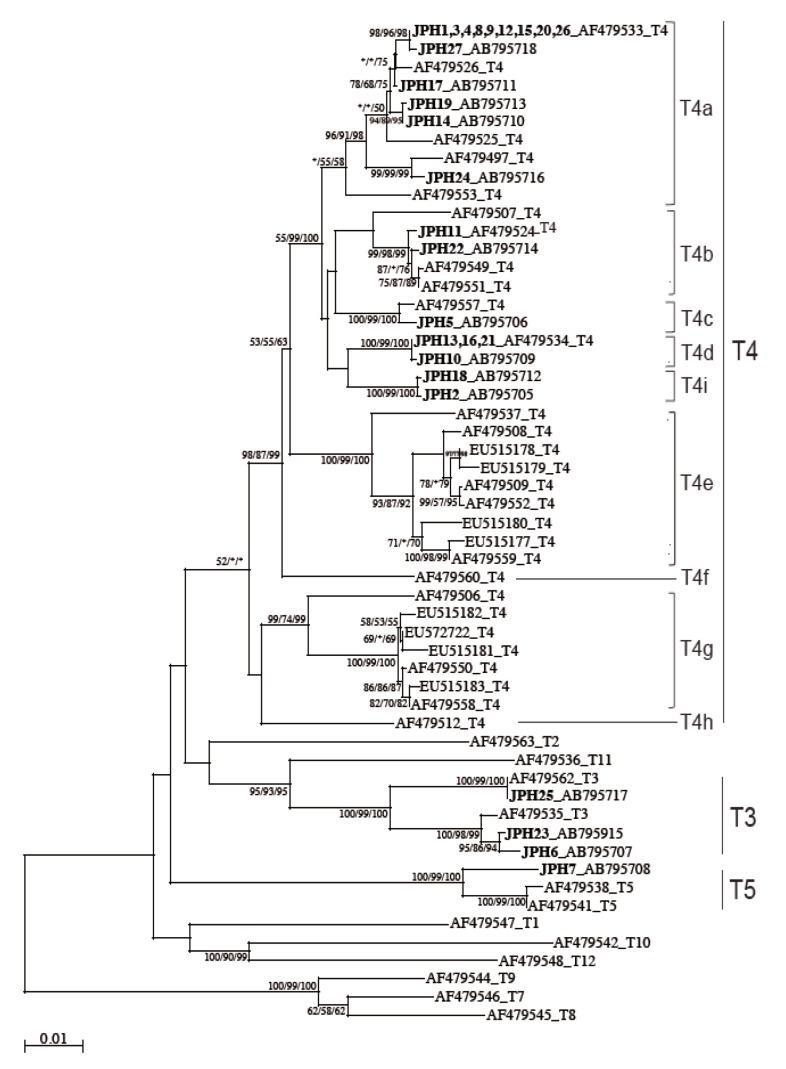
NJ tree reconstructed with the 16S rDNA sequences of Acanthamoeba. Isolates from Acanthamoeba keratitis are shown in boldface text, mixed infections with underlining, and with new or reference accession numbers. All reference sequences are shown with the accession numbers and the genotypes. Representative NJ tree with bootstrap value (1,000 replicates) for NJ, MP, and ML methods, conducted as described in "Materials and Methods", are shown. The analysis involved 55 nucleotide sequences. An asterisk indicates a value of less than 50% or if a position of the node is differ according to each analysis method. The evolutionary distances are shown in the units of the number of base substitutions per site.
Subcloning of genotyping results
Among the 27 strains, only 2, JPH5 and JPH17, showed mixed sequencing profiles (sequence heterozygosity at some nucleotide positions) of their 18S rRNA gene sequences. Specifically, among 7 clones isolated from JPH5, 4 and 3 clones were identified as JPH5A (U07408) and JPH5B (AY173000), respectively, whereas among 7 clones isolated from JPH17, 3 and 4 clones were identified as JPH17A (U07415) and JPH17B (U07403), respectively (Table 2; Fig. 2).

Comparison of genotyping results assessed in this study
Estimation of average evolutionary divergence of T4 genotypes
The numbers of base substitutions per site from averaging over all sequence pairs of the 18S rRNA gene and 16S rRNA gene were analyzed as described in "Materials and Methods". The confirmed average evolutionary divergences and standard errors of 18S rRNA gene and 16S rRNA gene were 0.010±0.003 and 0.013±0.003, respectively.
DISCUSSION
The increased risk of AK has been widely recognized worldwide, as well as in Japan. A recent survey conducted by the Japan Contact Lens Society and the Japanese Association for Ocular Infection in 224 facilities all over Japan from April 2007 to March 2009 revealed the high prevalence of AK in Japan. Specifically, among 350 patients who were diagnosed with contact lens-associated microbial keratitis, Acanthamoeba spp. were identified in 85 (24.3%) cases [24]. To date, the data regarding the distribution of various genotypes in Japan are still quite limited. Edagawa et al. [25] reported T4 isolates in tap-water samples, and multiple clinical T4 cases and 1 T11 case [26-27] have been reported so far.
In this study, 27 Acanthamoeba strains that caused AK in Japan were classified into 3 genotypes, T3 (3 strains), T4 (23 strains), and T5 (one strain) (Table 2; Figs. 2, 3), consistent with previous findings that both T3 and T4 genotypes were prevalent among AK patients around the world (Tables 4, 5). On the other hand, the T5 genotype was mostly detected in the environment [13,19,21,25], and the reports of the T5 genotype in AK patients [28,29] or human nasal mucosa [4,13] were very rare.

18S rRNA gene sequences used in this study
16S rRNA gene sequence used in this study
It seems interestingly, most of the haplotypes identified in this study showed 100% identity to the reference sequences available in the database. In the 18S rRNA gene analysis, only 5 were newly recognized haplotypes, and the rest of 22 haplotypes (including subcloned ones) had 13 homologous references; on the other hand, in the 16S rRNA gene analysis, 14 were newly identified, and the rest of 13 haplotypes had 3 references (Tables 4, 5). The places where the references were originally isolated include many areas all over the world, such as Africa, Argentina, France, Germany, India, Israel, Japan, Korea, Pakistan, Slovakia, Thailand, UK, and USA. Therefore, these Acanthamoeba haplotypes might have been distributing around the world and maintaining individual haplotypes independently, despite their geographically dispersed conditions. A robust feature of these haplotypes could be a confidential base for the molecular classification of Acanthamoeba spp.
The results of 18S and 16S rRNA genotyping were found to match perfectly with each other, and no contraindication depending on these loci was observed; however, the haplotype diversity of AK-related strains was clearly different between the 2 analyses (Figs. 2, 3). For example, while JPH3, 12, 17B, and 20 were identified as the same haplotype (U07403) by the 18S rRNA gene analysis, 9 strains (JPH1, 3, 4, 8, 9, 12, 15, 20, and 26) were identified as the same haplotype (AF479533) by the 16S rRNA gene analysis. Notably, JPH3, 12, and 20 were included in both analyses, but not other haplotypes. It is noteworthy that the genotyping assessments were conducted using gene segments of partial 18S rRNA (517-570 bp) and partial 16S rRNA (1,520-1,540 bp). That is, even with the use of longer nucleotide sequences, one third (9/27) of the strains were classified as 1 specific haplotype (AF479533) by the 16S rRNA gene analysis. The differences were statistically not significant; moreover, the average evolutionary divergence of all T4 samples confirmed in this study indicated higher values among 16S rRNA gene haplotypes (0.013±0.003) than 18S rRNA gene haplotypes (0.010±0.003), which may be consistent with a previous notion of the comparatively low divergence in the 18S rRNA gene locus [13,20,30-32]. While further research is required before a conclusive hypothesis can be drawn, we speculate that a part of T4 genotypes may be a dominant haplotype as a causal agent of AK and that the 16S rRNA gene analysis is useful for the evaluation.
Since the sub-genotype classification of the T4 cluster seems to be useful for higher-resolution molecular analyses [20,21,32], 8 subclusters (T4a-T4h) have been proposed [21]. In this study, the T4 sub-genotype analysis using the 16S rRNA gene locus revealed a clear sub-conformation within the T4 genotype and also leads to the recognition of a new sub-genotype, T4i, as the ninth sub-genotype in the T4 cluster (Fig. 3). We showed that the 2 newly identified sequences from JPH2 and JPH8 belonged to T4i, with statistically significant bootstrap values. Nevertheless, the number of genetic references for the 16S rRNA gene locus does not seem to be sufficient for completing the precise identification of all sub-genotypes. In the phylogenetic reconstruction (Fig. 3), some haplotypes, for example, AF479553 in T4a and AF479507 in T4b, were positioned as out-groups of the main cluster. These out-group haplotypes might be members of yet-to-be identified subclusters and could be categorized into novel clusters as the number of the reference sequences increases in the future.
Differences were observed between nuclear and mitochondrial genetic characteristics; that is, the mixed haplotype profiles from 2 individual strains, JPH5 and JPH17, were detected only in the nuclear gene analysis, but not in the mitochondrial gene analysis (Table 3; Figs. 2, 3). The formation of JPH5A/5B in the JPH5 strain and JPH17A/17B in the JPH17 strain seemed to be stable. To evaluate the contamination risk with other strains, we tried 1 trophozoite PCR after years of cultivation and got the same results for both strains (data not shown). The contamination might also be ruled out by the result of the 16S rRNA gene analysis, which showed only a single genotype for each strain.

18S rRNA sequence heterogeneities observed in two isolates of genotype T4
Taken these results together, these strains are considered to possess heterozygous nuclear 18S rRNA genes and homozygous mitochondrial 16S rRNA genes. The polyploid genome conformation of Acanthamoeba spp. has been suggested [33]; therefore, an accumulation of allelic sequence heterogeneity in polyploid genomes within a single cell might be a possible explanation for the mixed haplotype profile. However, such a possibility may be ruled out in our case, since all haplotypes identified in the sub-cloning analysis had their respective homologous haplotypes as individual strains. Specifically, JPH5A, 5B, 17A, and 17B were found to be identical to previous reported reference strains 82-12-324 (U07408), KA/MSS8-1 (AY173000), JAC/S2 (U07415), and V042 (U07403), respectively. Currently, there is no clear evidence for the meiotic or sexual process in the Acanthamoeba life cycle, even though the species apparently possesses a gene (Spo11) required for the meiotic recombination [34]. Whereas, the presence of heterozygous nuclear haplotypes in single strain, observed in this study, suggesting a possibility of the genetic hybridization between different strains of Acanthamoeba. Therefore, detailed evaluations of the presence of such mixed haplotypes in the population of Acanthamoeba spp. are considered to be required for the precise molecular taxonomy.
The nuclear 18S rRNA gene and mitochondrial 16S rRNA gene loci of Acanthamoeba spp. possess different and unique characteristics usable for the genotyping analyses, and those specific features could contribute to the establishment of molecular taxonomy for the species complex of Acanthamoeba.
ACKNOWLEDGMENTS
We would like to dedicate our work with our deepest empathy in the memories of Mr. Yoshizaki Kentaro who passed away before the completion of this study. This research was partly supported by Grants-in-Aid for Scientific Research from the Ministry of Environment, Japan. We would like to thank Editage for providing editorial assistance.