Rapid Detection and Identification of Wuchereria bancrofti, Brugia malayi, B. pahangi, and Dirofilaria immitis in Mosquito Vectors and Blood Samples by High Resolution Melting Real-Time PCR
Article information
Abstract
A simple, rapid, and high-throughput method for detection and identification of Wuchereria bancrofti, Brugia malayi, Brugia pahangi, and Dirofilaria immitis in mosquito vectors and blood samples was developed using a real-time PCR combined with high-resolution melting (HRM) analysis. Amplicons of the 4 filarial species were generated from 5S rRNA and spliced leader sequences by the real-time PCR and their melting temperatures were determined by the HRM method. Melting of amplicons from W. bancrofti, B. malayi, D. immitis, and B. pahangi peaked at 81.5±0.2℃, 79.0±0.3℃, 76.8±0.1℃, and 79.9±0.1℃, respectively. This assay is relatively cheap since it does not require synthesis of hybridization probes. Its sensitivity and specificity were 100%. It is a rapid and technically simple approach, and an important tool for population surveys as well as molecular xenomonitoring of parasites in vectors.
INTRODUCTION
Lymphatic filariasis, a mosquito-borne disease, is caused by filarial nematodes Wuchereria bancrofti, Brugia malayi, and Brugia timori [1]. It is a major public health problem, particularly in the tropics and subtropics, including Thailand and Myanmar [2,3]. Although it is not a major cause of mortality in humans, chronic pathology causes permanent disability and handicap. World Health Organization has categorized lymphatic filariasis as one of the 6 diseases which can be totally eliminated [4]. Several additional species of filarial nematodes, for example, Brugia pahangi and Dirofilaria immitis, infect various animals, such as dogs, cats, tigers, otters, tree shrews, and monkeys, in endemic areas of Southeast Asia, Malaysia, the Philippines, Indonesia, Pacific islands, and Thailand [5-7] and possibly cause human zoonotic diseases [8,9].
Control of lymphatic filariasis in human populations and zoonotic filariasis in feline and canine reservoirs should result in a decrease in human cases. The availability of effective methods to monitor the presence or absence of filarial larvae in the mosquito vector and/or microfilariae in the definitive hosts is important for interruption of the transmission of infection. However, microscopic methods for detection of filarial larvae in mosquito vectors and/or of microfilariae in the blood circulation are laborious and require highly experienced personnel, and still the sensitivity is not so high. Thus, a molecular detection tool is necessary to distinguish between closely related species or subspecies. Such tools include conventional PCR [10] and semi-nested PCR-RFLP [11]. However, these methods require analysis by agarose gel electrophoresis which is slow, having limited throughput and a bias toward carry-over contamination as well as spurious results. Recently, quantitative real-time PCR was reported to successfully detect W. bancrofti [12], B. malayi, B. pahangi [13,14], D. immitis [15], and Dirofilaria repens [16]. Also recently, a real-time PCR-based high-resolution melting (HRM) analysis was reported for detection and identification of B. malayi, B. pahangi, and D. immitis in blood samples in a single-tube assay [17]. This is a rapid and technically simple method which does not require fluorophore probes or oligonucleotides nor agarose gel electrophoresis [18]. However, a HRM-based real-time PCR for simultaneous detection of 4 filarial species, W. bancrofti, B. malayi, B. pahangi, and D. immitis has not been developed yet.
This study was conducted to develop a single tube, HRM-based real-time PCR for differentiation and detection of W. bancrofti, B. malayi, B. pahangi, and D. immitis in mosquito vectors and blood samples. The analytical sensitivity and specificity of the method were evaluated.
MATERIALS AND METHODS
Mosquitoes and experimental infections
Culex pipiens quinquefasciatus and Aedes aegypti mosquitoes were collected in urban areas of Khon Kaen Province, northeastern Thailand. Additionally, Aedes togoi was collected from Koh Nom Sao, Chanthaburi Province, eastern Thailand [19]. Cx. quinquefasciatus, Ae. togoi, and Ae. aegypti mosquitoes were artificially infected as previously described with W. bancrofti [12], B. malayi [20], and D. immitis [15], respectively.
Blood containing W. bancrofti microfilariae (density 44 microfilariae/20 µl) was collected from a male carrier, a Burmese immigrant, at Mae Sot District, Tak Province, northwestern Thailand. Blood containing B. malayi microfilariae (density 160 microfilariae/20 µl) was obtained from an experimentally infected cat. D. immitis-infected dog blood (density 54 microfilariae/20 µl) came from a veterinary clinic in Khon Kaen Province.
The mosquitoes were allowed to feed on heparinized blood using an artificial membrane feeding technique [21]. Fourteen days after feeding, each mosquito was dissected in normal saline solution, and the number of larvae was counted under a dissecting microscope. After dissection, all larvae, together with the insect's body, were mixed and put into a 1.5-ml microcentrifuge tube. Specimens were labeled and kept at -20℃ for DNA extraction.
This study was approved by the Human Ethics Committee of Khon Kaen University, based on the Ethics of Human Experimentation of the National Research Council of Thailand (Reference No. HE 42036).
Source of blood specimens and control DNAs for sensitivity and specificity
One W. bancrofti-infected blood sample was collected from an infected Burmese immigrant and 1 B. malayi-infected blood sample was collected from an experimentally infected cat (see above). A total of 14 DNA samples were extracted from EDTA blood samples of dogs obtained from the Animal Hospital of the Faculty of Veterinary Science, Chulalongkorn University, Bangkok, and from the Faculty of Veterinary Science, Khon Kaen University, Khon Kaen. Blood specimens were microscopically examined, and the presence of parasites was confirmed by staining a thick blood film (20 µl blood/slide) with Giemsa [22] and by special staining for acid phosphatase activity for differentiation of B. malayi and B. pahangi [23,24]. Out of these 14 DNA samples, 5 were from blood samples positive for D. immitis (range of microfilarial density=2-20/50 µl), 5 for B. pahangi (range of microfilarial density=1-78/50 µl), 2 for Hepatozoon canis and 2 for Babesia spp. DNAs extracted from blood samples from healthy dogs (n=5), a cat (n=1), and a human (n=1). DNAs from human red blood cells infected with Plasmodium falciparum (n=1) and P. vivax (n=1) were used for specificity evaluations.
For preparation of DNA specimens for real-time PCR, each mosquito specimen was put into a 1.5-ml microcentrifuge tube, homogenized with a disposable polypropylene pestle (BellCo Glass, Vineland, New Jersy, USA), and DNA extracted using a NucleoSpin Tissue Kit (Macherey-Nagel, Düren, Germany). The DNAs were eluted in 100 µl of 5 mM Tris-HCl, pH 8.5, and stored at -20℃ until use. A 5-µl sample of each DNA was used per reaction. Fifty-µl blood samples were used for DNA extraction and purification as described above.
HRM real-time PCR assay
LightCycler® PCR detection and analysis systems were used for amplification and quantification. Specific primers, i.e., SLX-F (5' GGAATCCCAGGTGTTGTAGACAT 3') and SLX-R (5' GGGCTGAAACATTCAATTACCTC 3') (Proligo, Helios, Singapore), were designed from B. malayi 5S rRNA gene and spliced leader sequence SL1 (GenBank accession no. D87037). According to bioinformatic analysis, the primers can anneal with Wuchereria bancrofti, Brugia malayi, Brugia pahangi, and Dirofilaria immitis genes, yielding amplicons of different sizes.
For amplification detection, a LightCycler 480 High Resolution Melting Master Kit (Roche Applied Science, Mannheim, Germany) was used as recommended by the manufacturer. The PCR mixture contained 1×LightCycler 480 HRM Master Mix, which included HRM dye, a DNA-binding dye which can be used at high concentrations without inhibiting PCR reactions, 2.2 mM MgCl2, 0.8 µM SLX-F primer, and 0.8 µM SLX-R primer. The total reaction volume was 20 µl. The PCR cycling for HRM curve acquisition was run under the following conditions: 1 cycle pre-incubation 10 min at 95℃; 45 cycles of 95℃ for 10 sec, 55℃ for 8 sec, and 72℃ for 15 sec. After amplification, the reaction product was melted by raising the temperature from 60℃ to 90℃, with an increment of 0.1℃/sec, to obtain information on melting profiles. Products were then cooled to 40℃ for 30 sec. All samples were tested in duplicate in 96-well plates.
In order to determine the specificity of the HRM real-time PCR assay, DNAs extracted from samples (see above) were analyzed separately. Each run contained at least 1 negative control (consisting of 5 µl distilled water) and 1 positive control (plasmid in water).
Melting curve analysis to determine melting temperature (Tm) of the specific PCR product, of which identification was confirmed by conventional gel electrophoresis, was performed using LightCycler 480 gene scanning software (version 1.5) (Roche Applied Science). The cycle number indicating the target sequence copy number was presented as the number of PCR cycles needed for the change in fluorescence signal of the amplicons to exceed the detected threshold value.
Positive control plasmids
A positive control plasmid was constructed for each worm species by cloning a PCR product amplified by the SLX-F and SLX-R primers into the pGEM-T Easy plasmid (Promega, Madison,Wisconsin, USA), according to the manufacturer's instructions. Each plasmid was propagated in Escherichia coli JM109. Each inserted gene was sequenced in both directions; the resulting sequences were identical with the gene sequences from which the PCR primers were designed.
RESULTS
Standardization of HRM real-time PCR
The sensitivity of HRM real-time PCR was determined using 10-fold serial dilutions of W. bancrofti-positive control plasmid DNA (9 to 9×107 copies), B. malayi-positive control plasmid DNA (2 to 2×107 copies), D. immitis-positive control plasmid DNA (1 to 107 copies) and B. pahangi-positive control plasmid DNA (4 to 4×107 copies) in water. At a cut-off at 40 PCR cycles, the limit of detection for W. bancrofti was ≤9 copies of positive control (Fig. 1A), for B. malayi the limit was ≤2 copies of positive control (Fig. 1B), for D. immitis the limit was 10 copies of positive control (Fig. 1C), and for B. pahangi the limit was ≤4 copies of positive control (Fig. 1D). No fluorescence was detected when testing purified DNA from non-infected mosquitoes (Cx. quinquefasciatus and Ae. aegypti), human, dog, and cat blood samples, dog blood samples infected with H. canis and Babesia spp. or P. falciparum, and P. vivax-infected human red blood cells.
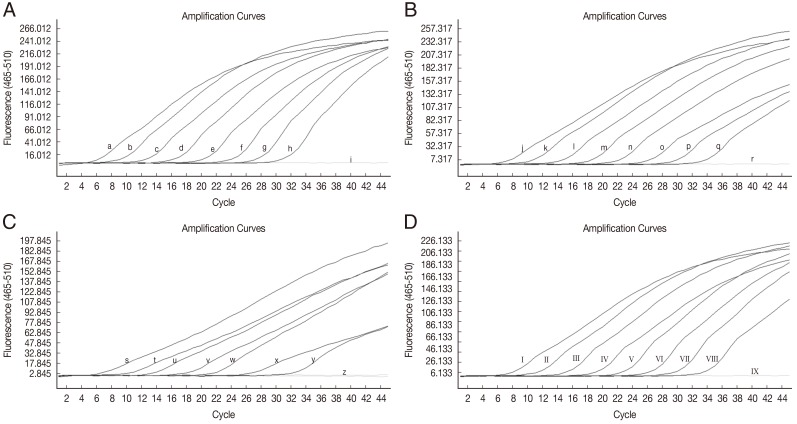
Amplification plot of fluorescence (y-axis) vs cycle numbers (x-axis) showing the analytical sensitivity of HRM real-time PCR for detecting DNA of W. bancrofti (A), B. malayi (B), D. immitis (C), and B. pahangi (D) in control plasmids. (a-h) 10-fold dilutions of W. bancrofti plasmid from 9×107 to 9 copies/reaction (i) distilled water. (j-q) 10-fold dilutions of B.malayi plasmid from 2×107 to 2 copies/reaction (r) distilled water. (s-y) 10-fold dilutions of D. immitis plasmid from 107 to 10 copies/reaction (z) D. immitis plasmid 1 copy/reaction and distilled water. (I-VIII) 10-fold dilutions of B. pahangi plasmid from 4×107 to 4 copies/reaction (IX) distilled water.
HRM real-time PCR for detection of filarial nematodes in mosquitoes and blood samples
HRM real-time PCR was applied to detect W. bancrofti, B. malayi, and D. immitis in infected mosquitoes and W. bancrofti, B. malayi, D. immitis, and B. pahangi in blood samples (Table 1). DNAs from uninfected dog blood samples and blood samples infected with other pathogens were not amplified by this primer set. The results of melting curve analyses are given in Fig. 2 and Table 1. Furthermore, the HRM real-time PCR could detect as little as a single L3 in mosquitoes infected with W. bancrofti, B. malayi, or D. immitis, or a single microfilaria of W. bancrofti, B. malayi, B. pahangi, and D. immitis per 50-µl blood sample. The sizes of the amplified products are 227 bp for W. bancrofti, 199 bp for B. malayi, 201 bp for B. pahangi, and 161 bp for D. immitis. The sensitivity and specificity were both 100%.

HRM real-time PCR showing cycle number (Cn) and melting temperatures (Tm) for the detection of Wuchereria bancrofti, Brugia malayi, Dirofilaria immitis, and Brugia pahangi in infected mosquitoes and blood samples

Representative melting curve analyses of the amplification products of W. bancrofti, B. malayi, D. immitis, and B. pahangi 5S rRNA gene and spliced leader sequence. The figure shows the melting curves of D. immitis plasmid DNA (106 copies) (a), D. immitis-infected blood sample (b), B. malayi plasmid DNA (2×104 copies) (c), B. malayi-infected blood sample (d), B. pahangi plasmid (4×104 copies) (e), B. pahangi-infected blood sample (f), W. bancrofti plasmid (9×104 copies) (g), W. bancrofti-infected blood sample (h), non-infected Ae. aegypti and Cx. quinquefasciatus, P. falciparum-infected and P. vivax-infected human red blood cells, non-infected cat, human, and dog blood samples, Babesia spp., and H. canis-infected dog blood DNA, respectively, and distilled water (all i).
DISCUSSION
The HRM real-time PCR assay has been used for simultaneous discrimination between B. malayi and B. pahangi in cat reservoirs [25] and recently, to distinguish between B. malayi, B. pahangi, and D. immitis microfilariae in blood samples by different Tm [17]. W. bancrofti is still a major debilitating and disfiguring disease. It is considered that many tropical countries are at risk of acquiring the infection [26]. In Thailand, bancroftian filariasis is endemic in the Thai-Myanmar border regions [3]. This prompted us to develop the HRM real-time PCR assay for differential detection of 4 filarial species; W. bancrofti, B. malayi, D. immitis, and B. pahangi in mosquito vectors and blood samples. The HRM assay could detect the 5S rRNA gene and spliced leader sequences by using a single primer pair that can amplify products from all 4 species. The assay could distinguish each species according to its melting temperature based on different GC content and the length of the amplicon [27]. No PCR products were amplified from a wide range of control DNAs from other parasites or from intermediate and definitive hosts, indicating high specificity.
On a cost-per-sample basis, this method is cheaper than using multiplex real-time PCR methods that require synthesis of specific labeled probes. The assay can be performed on a large number of samples at the same time, with only very small sample volumes required.
In conclusion, a rapid, sensitive, and specific HRM real-time PCR for the detection of 4 filarial nematodes in mosquito vectors and blood samples is presented. This procedure is a suitable tool not only for diagnosis and epidemiological surveys of filariasis and dirofilariasis in final hosts but also for molecular xenomonitoring of W. bancrofti, B. malayi, B. pahangi, and D. immitis in mosquito vectors. As D. repens has also been reported as a causative agent of human filariasis in Thailand [28], we intend to seek material of this species to extend the experimental work reported here.
ACKNOWLEDGMENTS
This research was supported by the National Science and Technology Development Agency (Discovery Based Development Grant); the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission through the Health Cluster (SHeP-GMS) and Khon Kaen University, Thailand. Wanchai Maleewong, Pewpan M. Intapan, and Tongjit Thanchomnang are supported by TRF Senior Research Scholar Grant, Thailand Research Fund grant no. RTA5580004.