Cryptosporidium spp., Giardia intestinalis, and Enterocytozoon bieneusi in Captive Non-Human Primates in Qinling Mountains
Article information
Abstract
Non-human primates (NHPs) are confirmed as reservoirs of Cryptosporidium spp., Giardia intestinalis, and Enterocytozoon bieneusi. In this study, 197 fresh fecal samples from 8 NHP species in Qinling Mountains, northwestern China, were collected and examined using multilocus sequence typing (MLST) method. The results showed that 35 (17.8%) samples were positive for tested parasites, including Cryptosporidium spp. (3.0%), G. intestinalis (2.0%), and E. bieneusi (12.7%). Cryptosporidium spp. were detected in 6 fecal samples of Macaca mulatta, and were identified as C. parvum (n=1) and C. andersoni (n=5). Subtyping analysis showed Cryptosporidium spp. belonged to the C. andersoni MLST subtype (A4, A4, A4, and A1) and C. parvum 60 kDa glycoprotein (gp60) subtype IId A15G2R1. G. intestinalis assemblage E was detected in 3 M. mulatta and 1 Saimiri sciureus. Intra-variations were observed at the triose phosphate isomerase (tpi), beta giardin (bg), and glutamate dehydrogenase (gdh) loci, with 3, 1, and 2 new subtypes found in respective locus. E. bieneusi was found in Cercopithecus neglectus (25.0%), Papio hamadrayas (16.7%), M. mulatta (16.3%), S. sciureus (10%), and Rhinopithecus roxellana (9.5%), with 5 ribosomal internal transcribed spacer (ITS) genotypes: 2 known genotypes (D and BEB6) and 3 novel genotypes (MH, XH, and BSH). These findings indicated the presence of zoonotic potential of Cryptosporidium spp. and E. bieneusi in NHPs in Qinling Mountains. This is the first report of C. andersoni in NHPs. The present study provided basic information for control of cryptosporidiosis, giardiasis, and microsporidiosis in human and animals in this area.
INTRODUCTION
Cryptosporidiosis, giardiasis, and microsporidiosis, 3 emerging infectious diseases caused by Cryptosporidium, Giardia, and microsporidia in humans and animals, have been reported both in developed countries and in those under development in last decades [1-3]. Clinical signs of these diseases are normally chronic diarrhea in immune-competent hosts, but these infections may lead to significant morbidity and mortality in immune-compromised or -suppressed hosts, such as AIDS patients [1,4].
Accurate identification of species/genotype/subtype is essential for characterization of transmission pattern and control of parasitic diseases. Until now, there are 27 valid species of Cryptosporidium and over 70 genotypes have been described in the literature, and at least 14 species and 5 genotypes were detected in humans [3,5]. More than 50 species of Giardia have been proposed during 1920-1930, but only 6 species are accepted by most researchers on the basis of the morphology of the trophozoites/cysts [4]. Giardia intestinalis (syn. G. lamblia, G. duodenalis), the only species that can infect humans, contains at least 7 valid assemblages (A-G) within species. The assemblages A and B are responsible for most of human infections [4]. Likewise, some Enterocytozoon bieneusi genotypes are mostly host-adapted in nature, and some are zoonotic [2]. Epidemiological surveys indicated that humans acquire infections of Cryptosporidium spp., G. intestinalis, and E. bieneusi mainly through the fecal-oral route via direct person-to-person or animal-to-person contact, or ingestion of contaminated water or food by animal manure [1,4]. Zoonotic Cryptosporidium spp., G. intestinalis, and E. bieneusi have been detected in domestic livestock, companion and wild animals. Their hosts have been believed to play significant roles in this context, contributing to parasite oocysts/cysts in large proportion [1,2,4].
Data from several studies suggested that non-human primates (NHPs) might be sources of cryptosporidiosis, giardiasis, and microsporidiosis for human infections [6-11], since they have close relationships with human and can harbor zoonotic species/genotypes/subtypes. The infections of human-pathogenic Cryptosporidium spp., G. intestinalis, and E. bieneusi in NHPs have been documented in Asia, America, and Africa [6-11]. In Uganda, potential cross-transmissions between human and mountain gorillas for G. intestinalis assemblage A and C. parvum were reported [6]. In China, the respective prevalences of human-pathogenic Cryptosporidium spp. and G. intestinalis infections were 10.9% and 8.3%, respectively, in Macaca mulatta from Guizhou [7]. The zoonotic C. hominis and G. intestinalis were also detected in M. fascicularis from a commercial animal facility in Guangxi of China [8]. E. bieneusi was reported in 5 NHP species from Guangdong, Yunnan, Guangxi, Henan, and Sichuan provinces, including M. fascicularis (67.7%), M. mulatta (8.8%), M. fuscata (33.3%), Trachypithecus leucocephalus (13.6%), and Rhinopithecus roxellana (9.5%) [9].
Qinling Mountains, the natural boundary between the North and South of China, are mainly situated in Shaanxi province. The mountains support huge varieties of endangered wild and captive animals, such as R. roxellana, Budorcas taxicolor, Nipponia nippon, and Ailuropoda melanoleuca. Here, 2 largest captive animal sites in Shaanxi province at the foot of Qinling Mountains were selected to determine the prevalence and species/genotype/subtypes distribution of Cryptosporidium spp., G. intestinalis and E. bieneusi in captive NHPs, and to unravel their public health significance.
MATERIALS AND METHODS
From December 2013 to July 2014, 197 fresh fecal specimens from 8 species of NHPs were collected from 2 largest captive wild animal farms in Qinling Mountains (Table 1) according to the legal requirements of guide for the Care and Use of Laboratory Animals of the Ministry of Health, China, and approval of the Research Ethics Committee of Northwest A&F University (No. NWAFREC-2013-09-0002). One farm covers over 173 ha and houses 10,000 wild animals belonging to almost 300 species comprising mammals, birds, and reptiles. The other (5.3 ha) harbors 600 animals which were rescued from Qinling Mountains. Each site is near to human habitats and is visited by more than 100 million or 50 million individuals each year. Visitors are allowed to watch them from a short distance. All NHP species of 2 origins are kept in mono-specific groups sharing each respective pens/cages. All NHPs are housed in cages/pens littered with natural materials such as the ground bark. Fresh stool samples from the rectum of each of rare NHPs were collected, placed immediately in disposable plastic bags, and stored at 4˚C for further study. All animals in this study showed no clinical symptoms.

Distribution of Cryptosporidium spp., G. intestinalis and E. bieneusi in non-human primates (NHPs) in Qinling Mountains, northwestern China
Genomic DNA was extracted from each fecal specimen using the E.Z.N.A® Stool DNA Kit (OMEGA Bio-tek, Inc, Georgia, USA) according to manufacture-recommended procedures, and stored at -20˚C for molecular analysis. The gene loci of triose phosphate isomerase (tpi), small subunit ribosomal RNA (SSU rRNA), beta giardin (bg), and glutamate dehydrogenase (gdh) loci were used to determine G. intestinalis assemblages according to protocols described previously [10]. The 18S rRNA and Cryptosporidium oocyst wall protein (COWP) genes were used to indicate Cryptosporidium species/genotypes [10,12]. The 60 kDa glycoprotein (gp60) gene was employed specifically for detection and assignment of Cryptosporidium species/genotypes and subtypes that are infective to humans [13]. To further analyze C. andersoni subtypes, 4 minisatellite loci (CM-MS1, CM-MS2, CM-MS3, and CM-MS16) were used [12]. A 392 bp fragment of the internal transcribed spacer (ITS) gene was amplified by nested PCR as previously to genotype E. bieneusi [9].
The TaKaRa Ex Taq (TaKaRa Shuzo Co., Ltd., Otsu, Japan) was used to amplify all target loci. The PCR reactions were conducted in 25 μl reaction mixtures containing 1×Ex Taq buffer (Mg2+ free), 2 mM MgCl2, 0.2 mM each deoxy-ribonucleoside triphosphate (dNTP), 0.4 μM each primer, 0.625 U TaKaRa ExTaq DNA polymerase, and 2 μl of DNA template (or 1 μl of the first PCR amplicon). For the amplication of SSU rRNA for G. intestinalis, 1 μl dimethyl sulfoxide (DMSO) was added additionally. Replicate analysis was done at each locus with both positive and negative controls. The secondary PCR products were visualized on a UV transilluminator after electrophoresis in 1.5% agarose gel and stained with ethidium bromide to validate amplification efficiency.
All positive PCR products were sequenced directly by Shanghai Sangon Biological Engineering Biotechnology Company using ABI 3730xl automated DNA sequencer (Big Dye Terminator Chemistry, Applied Biosystems, Foster City, California, USA). The species/assemblages/genotypes/subtypes of enteric parasites detected were determined by the alignment with reference sequences from the GenBankTM database using BLAST program (http://www.ncbi.nlm.nih.gov). Phylogenetic trees were implemented in the program Mega 5.0 (http://www.megasoftware.net/) using a neighbor-joining method. Bootstrap analysis was used to assess the robustness of clusters with 1,000 replicates.
RESULTS
Of 197 samples examined, 35 (17.8%) were positive for tested parasites. The differences in prevalence among infections of different parasites in NHPs were compared using the χ2 test in SPSS 17.0 (http://www-01.ibm.com/software/analytics/spss/), with statistically significant at P<0.05. The differences of infection rates for 3 parasites were not statistically significant (P>0.05). Cryptosporidium spp. were detected in 6 fecal samples from M. mulatta (7.0%), with the overall infection rate of 3.0% in NHPs (Table 1), which was higher than that reported in laboratory M. fascicularis (0.5%) and NHPs (0.7%) in China, newly captive Papio anubis (2.6%) in Kenya, but lower than that in M. mulatta (10.9%) in China, P. anubis (11.9%) and Cercopithecus aethiops (29.3%) of Ethiopia [7,10,14,15].
G. intestinalis was identified in 4 specimens of M. mulatta (3.5%) and S. sciureus (5.0%), with the overall prevalence in NHPs in this study was 2.0% (Table 1). Different infection rates of G. intestinalis have been reported in different NHPs in China and other countries: 47% in Lemur catta in Italy, 18% in NHPs in Brazil, 9% in mountain gorillas in Rwanda, 8.5% in M. mulatta, and 2.2% in NHPs in China [10,16-18].
E. bieneusi was detected in 25 (12.7%) specimens from 5 captive NHP species, including C. neglectus (25.0%), P. hamadrayas (16.7%), M. mulatta (16.3%), S. sciureus (10.0%), and R. roxellana (9.5%) (Table 1). Recently, E. bieneusi frequently presented in P. troglodytesin (33.3%) in the Slovak Republic, captive P. anubis (12.3%) and P. troglodytesin (2.6%) in Kenya, M. fascicularis (18.5%) and NHPs (11.4%) in China, and P. troglodytesin (4.5%) in Cameroon [8,15,19]. The differences of infection status of these parasites may be related to the species of NHP, sample sizes, examination methods, different management systems, the timing of specimen collection, and geo-ecological conditions.
DISCUSSION
In the present study, of all pathogens, single infection of C. andersoni, C. parvum, G. intestinalis, and E. bieneusi was presented in fecal samples of M. mulatta, S. sciureus, C. neglectus, P. hamadrayas, and R. roxellana. Double infections of C. andersoni and E. bieneusi were co-occurred only in 4 samples of M. mulatta, but no other mixed infections were observed. These results suggested that single pathogen infection was more common in NHPs in this area.
Previous studies indicated that 5 Cryptosporidium species, namely C. hominis, C. parvum, C. felis, C. muris, and C. ubiquitum, have been detected in NHPs [1,7,10]. In the present study, sequence analysis revealed the presence of 2 Cryptosporidium species, namely C. andersoni (n=5, GeneBank accession no. KJ917574 to KJ917578, KJ917580 to KJ917584) and C. parvum (n=1, KJ917579, KJ917585) in M. mulatta (Table 1). The successful amplification of gp60 gene was only observed for the fecal sample with the infection of C. parvum. BLSAT search and alignment of the obtained gp60 gene sequence (KJ917586) from M. mulatta had 100% identity with the isolates Cp12 (GU214367) and D536 (FJ897784) from diarrheal patients in England and India [20,21], which indicated C. parvum isolate in this study belonged to the subtype IId A15G2R1 (KJ917586) based on the “genotypic” nomenclature described previously [20]. Based on gp60, subtype families Ia, Ib, Id, Ie, If, and Ii of C. hominis and IIc of C. parvum have been reported in NHPs [7,10,20]. In China, the subtype IId of C. parvum was detected in pre-weaned dairy cattle, hamsters, urban wastewater, and HIV-positive patients [22]. In the present study, 1 C. parvum gp60 subtype IId A15G2R1 was identified in M. mulatta and identical to the isolate from human patients in England and India [20,21]. This is the first report of subtype IId in NHPs, and it suggested that NHPs in Qinling Mountains may be potentially responsible for some zoonotic transmission of C. parvum.
C. andersoni, the common species in ruminants, was detected in this study. The zoonotic potential of C. andersoni was controversial in previous studies. The presence of partial 18S rRNA that very likely represented C. andersoni was confirmed in a male HIV-positive patient in France [23], and C. andersoni was detected by PCR/RFLP and DNA sequencing of 18S rRNA and COWP genes in 3 diarrheal patients in England between 1985 and 2000 [24]. However, no shedding oocysts were observed in M. fascicularis under either both normal or immunosuppressive conditions by orally inoculated with oocysts of 2 different C. andersoni Kawatabi types and C. muris RN-66 [25]. Herein, Cryptosporidium oocysts from M. mulatta with average size of 5.62×7.36 μm were observed under optical microscope using modified acid-fast technique, and it was identified as C. andersoni by sequencing of 18S rRNA (KJ917574 to KJ917578) and COWP (KJ917580 to KJ917584) genes. To subtype C. andersoni isolates in this study, each isolate of C. andersoni was successfully amplified and sequenced at 4 loci. Sequence analysis (KJ917587 to KJ917606) indicated haplotypes A4, A4, A4, and A1 at loci MS1, MS2, MS3, and MS16, respectively, forming 1 MLST subtype A4, A4, A4, and A1, which was identical to C. andersoni isolates from cattle in Henan, Jilin, Guangxi, Heilongjiang, Sichuan, and Shaanxi of China [12]. These findings may indicate C. andersoni infecting certain species of NHPs, such as M. mulatta, or distinct biological characteristics of different C. andersoni isolates. Therefore, further researches on animal adaptation and infectivity of C. andersoni obtained will be studied to illuminate zoonotic potential of C. andersoni in the future.
Of 8 G. intestinalis assemblages, assemblages A and B were commonly detected in NHPs, with the assemblage B dominating [4,10,26]. The assemblage E, restricted to ruminants, was also found in Ptephrosceles tephrosceles in western Uganda by real-time PCR and sequence analysis of ef1-a, SSU rRNA, tpi, and gdh genes, which suggested that livestock-to-primate transmission was most likely in assemblage E [27]. In the present study, 4 G. intestinalis-positive samples in M. mulatta (n=3) and S. sciureus (n=1) were successfully sequenced at 4 gene loci (KJ917607 to KJ917622) (Table 1). BLAST search against NCBI nucleotide sequence database showed that G. intestinalis isolates in this study were all belonged to G. intestinalis assemblage E. The reasons may be due to the fact of assemblage E being predominant in NHPs here, or small numbers of NHPs investigated. Considering a study based on PCR and DNA sequence analysis of tpi gene showed the assemblage E as the second dominating assemblage in human in Egypt [26], the NHPs in this study may have zoonotic potential for transmission of G. intestinalis.
Sequence analysis indicated no sequence differences of SSU rRNA gene (KJ917607 to KJ917610) among 4 G. intestinalis-positive samples, with 99% identity to G. intestinalis assemblage E isolates from cattle (KF843921) and goats (AY826210). The intra-genotypic diversity of 4 G. intestinalis isolates was observed at loci of tpi (KJ917619 to KJ917622), bg (KJ917611 to KJ917614), and gdh (KJ917615 to KJ917618) genes (Table 2). Four single nucleotide polymorphisms (SNPs) were detected at the tpi locus, forming 3 new assemblage E subtypes (KJ917619 to KJ917622). There were 2 SNPs among bg gene sequences, forming 1 new subtype (KJ917611, KJ917613) and 1 known subtype reported in Bison bonasus (FJ472822). Three subtypes were detected at the gdh gene locus, with 2 new subtypes (KJ917615, KJ917616) and 1 same to isolates Ly20 from cattle (KF843923). These results indicated complicate variability of G. intestinalis assemblage E isolates from different hosts and origins. One subtype in each of bg and gdh loci in this study had 100% identity with isolates from bovine in China (KF843923, FJ472822), which suggested animal-to-animal transmission may exist for G. intestinalis.

Variations in the nucleotide sequences of tpi, gdh and bg genes among subtypes of G. intestinalis assemblage E
In the present study, 25 specimens were amplified successfully at the ITS region of E. bieneusi. Sequence analysis indicated that these samples belonged to 5 distinct genotypes, including genotypes D (n=10, KM591953 to KM591962), BEB6 (n=4, KM591947 to KM591950) and 3 novel genotypes, namely MH (n=7, KM591963 to KM591969), XH (n=2, KM591951, KM591952) and BSH (n=2, KM591945, KM591946). The genotype D is the dominating genotype, and has been detected in M. mulatta (n=6), R. roxellana (n=2), and P. hamadrayas (n=2). The genotype BEB6 previously described in cat and sheep [2,11] was found in 4 fecal samples from R. roxellana. The novel genotype MH was found only in M. mulatta (n=7), with 2 SNPs to genotype D (GQ406055). Phylogenetic analysis (Fig. 1) showed that the novel genotype MH was closely related to genotype D and clustered in Group 1a that was regarded as zoonotic previously [2]. Considering that the genotypes D has been reported in humans, NHPs and urban wastewater [9,28,29], NHPs in Qinling Mountains would have risk for zoonotic transmission potential. The new genotypes XH and BSH were observed in respective fecal samples from S. sciureu (n=2) and C. neglectus (n=2). The genotype BSH contained 6 SNPs compared with BEB6 (KJ668737) grouped into genotypes of Group 2 reported only in animals [2]. Sequence comparisons revealed that the sequences of genotypes XH had 4 SNPs compared to genotypes CD5 (KJ668732) and CM4 (KF543866). In the phylogenetic tree, the novel genotype XH clustered with genotype CD5 from dog and genotype CM4 from NHPs in China [10,28], and this cluster was located in a solitary clade and was proposed as Group 9 (Fig. 1).
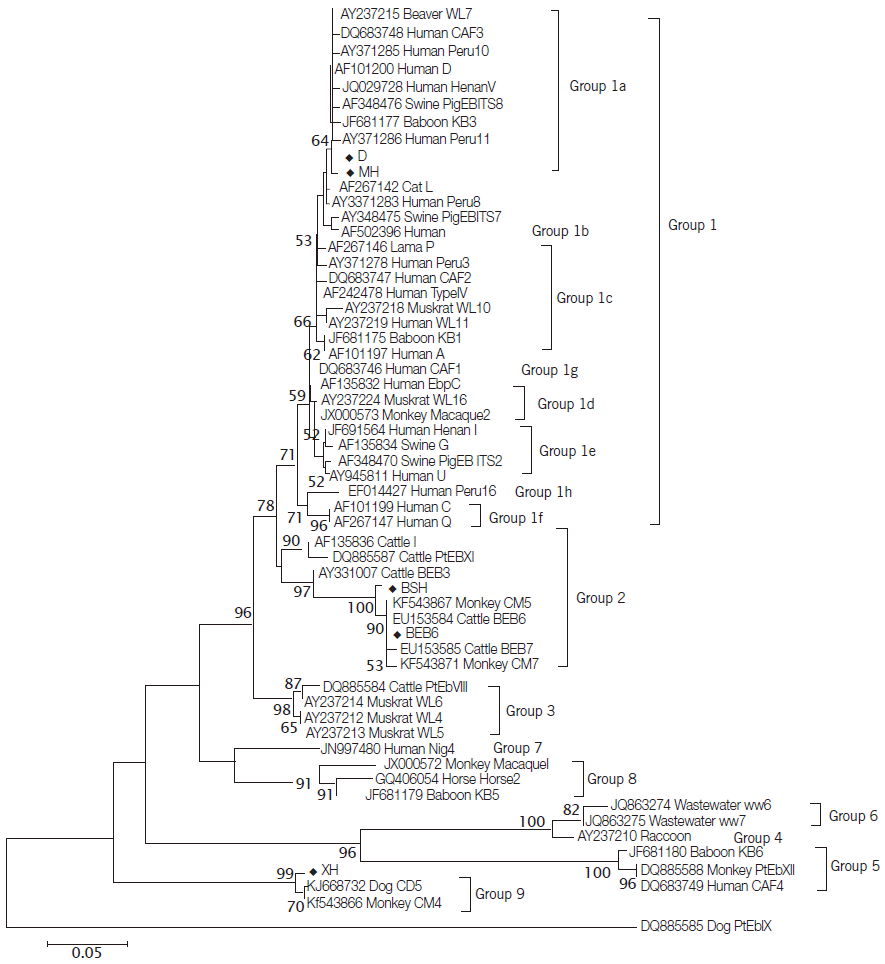
Phylogenetic relationships of E. bieneusi genotypes identified in this study and other genotypes previously deposited in GenBank as inferred by the neighbor-joining analysis of ITS sequences based on genetic distances calculated by the Kimura 2-parameter model. Bootstrap values greater than 50% from 1,000 replicates are shown on nodes. Each sequence from GenBank is identified by the accession number, host origin, and the genotype designation. Genotypes identified in this study are indicated by the symbol (◆).
In conclusion, the infections of Cryptosporidium spp., G. intestinalis, and E. bieneusi were detected in captive NHPs in Qinling Mountains. C. andersoni and C. parvum subtype IId A15G2R1 were firstly reported in NHPs. The assemblage E was the only G. intestinalis assemblage detected in NHPs in this study, and new subtypes at 4 loci were also found. Zoonotic E. bieneusi genotypes D and host-adapted genotype BEB6 were both presented in NHPs, and 3 novel genotypes were found. These results suggested that captive NHPs in Qinling Mountains would have significant zoonotic potential. Therefore, integrity measure should be implemented to control infections of Cryptosporidium spp., G. intestinalis and E. bieneusi in NHPs, and special care should also be taken by the animal attendants, animal care specialists, veterinarians, scientists and visitors to zoonotic transmission of cryptosporidiosis, giardiasis and microsporidiosis.
Acknowledgements
This study was supported by grants from the Program for New Century Excellent Talents in University (grant no. NCET-13-0489), the State Key Program of National Natural Science Foundation of China (grant no. 31330079), the Fund for Basic Scientific Research (grant no. ZD2012010), and the Open Funds of the State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (grant no. SKLVEB2013KFKT007).
Notes
The authors declare that they have no competing interests.