Complete Mitochondrial Genome of Echinostoma hortense (Digenea: Echinostomatidae)
Article information
Abstract
Echinostoma hortense (Digenea: Echinostomatidae) is one of the intestinal flukes with medical importance in humans. However, the mitochondrial (mt) genome of this fluke has not been known yet. The present study has determined the complete mt genome sequences of E. hortense and assessed the phylogenetic relationships with other digenean species for which the complete mt genome sequences are available in GenBank using concatenated amino acid sequences inferred from 12 protein-coding genes. The mt genome of E. hortense contained 12 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and 1 non-coding region. The length of the mt genome of E. hortense was 14,994 bp, which was somewhat smaller than those of other trematode species. Phylogenetic analyses based on concatenated nucleotide sequence datasets for all 12 protein-coding genes using maximum parsimony (MP) method showed that E. hortense and Hypoderaeum conoideum gathered together, and they were closer to each other than to Fasciolidae and other echinostomatid trematodes. The availability of the complete mt genome sequences of E. hortense provides important genetic markers for diagnostics, population genetics, and evolutionary studies of digeneans.
INTRODUCTION
About 500 species of echinostomatid flukes (Echinostomatidae) had been reported in the world [1], and at least 20 species belonging to 8 genera can infect humans [2]. Among these genera, Echinostoma is the largest one, which constitutes 7 species, including E. hortense, E. angustitestis, E. cinetorchis, E. echinatum, E. ilocanum, E. macrorchis, and E. revolutum [2]. Although the majority of people infected by echinostomes have no obvious symptoms, severe infections may cause anorexia, lower extremity edema, anemia, weight loss, and dysplasia [3].
The adult worms of E. hortense often inhabit the small intestine of humans and many animals [3]. The parasitic fluke E. hortense was originally described from rats in Japan [4], and since then, it has been reported in South Korea and China [2,3,5,6]. In addition to rats, it has also been found from cats, dogs, and pigs [6-8], and more importantly, there are many infected human cases in China, Japan, and South Korea [2,3,9-11]. A high prevalence of human infection by E. hortense was reported in Koje-myon, Kochang-gun, Kyongsangnam-do (Province), South Korea, up to 9.5% [11].
Mitochondrial (mt) genome sequences provide valuable genetic markers for investigating population genetics, systematics, and phylogenetics of animal species [12,13]. So far, there are about 18,000 species of digenean trematodes found in the world [14], but only 32 species have complete mt genomes sequenced. In the family Echinostomatidae, only 1 species Hypoderaeum conoideum (KM111525) had complete mt genome reported, and no mt genome of genus Echinostoma has been available. Previous studies on E. hortense have mainly focused on their morphology, life history, and epidemiological investigations [3-11]. However, the mitochondrial genome of this fluke has not been known yet. Therefore, in the present study, we have determined the complete mt genome sequences of E. hortense and inferred its phylogenetic relationship with other digenean trematodes.
MATERIALS AND METHODS
Parasites and total genomic DNA isolation
Adult E. hortense worms were collected from the small intestine of a naturally infected dog in Daqing, Heilongjiang Province of China. The trematodes were washed in physiological saline, identified morphologically as E. hortense based on existing keys and descriptions [15], fixed in 70% (v/v) ethanol (Yixin Biological Technology Co., Shanghai, China), and stored at -20˚C until use. Total genomic DNA was extracted from individual fluke using a TIANamp Genomic DNA Kit (TIANGEN, Beijing, China) according to the manufacturer’s instructions, and DNA sample was stored at -20˚C until use. In order to independently verify the identity of the specimens, the internal transcribed spacer 2 (ITS2) was amplified from the extracted genomic DNA by PCR and sequenced according to the conventional method. The ITS2 sequences obtained were perfectly matched with that of E. hortense available in GenBank (U58101.1).
Amplification, sequencing, and assembling of mt DNA fragments
Amplification, sequencing, and assembly of mtDNA fragments were performed according to the methods previously described [12,16]. The 7 pairs of oligonucleotide primers were designed based on the conserved regions from published complete mtDNA sequences of Fasciola hepatica, Opisthorchis felineus, Paramphistomum cervi, and H. conoideum (Table 1). The cycling conditions were 94˚C for 5 min (initial denaturation); then 94˚C for 1 min (denaturation), 50˚C for 1 min (annealing), 72˚C for 1-3 min (extension) for 35 cycles, and a final extension at 72˚C for 7 min. Each PCR reaction yielded a single band, detected in a 1.0% (w/v) agarose gel stained with ethidium-bromide. PCR products were directly sequenced on an ABI 3370 DNA sequencer at Shanghai Sangon Biotech Company (Shanghai, China) using a primer walking strategy. The complete mtDNA sequences of E. hortense were assembled using DNAStar software as a sequence editor [17].

Sequences of primers used to amplify PCR fragments of mitochondrial genome from Echinostoma hortense
Sequence analysis of E. hortense mt genome
Gene annotation, genome organization, translation initiation, translation termination codons, and the boundaries between protein-coding genes of mtDNA were identified based on comparison with mt genomes of other trematodes reported previously [18]. Open reading frames and gene boundaries were confirmed by comparing with H. conoideum mt genome nucleotide sequences (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Translation initiation and translation termination codons were identified using genetic codon table for mitochondrion in MEGA 5 [19] and also based on comparison with the mt genomes of the trematodes reported previously [16,18]. The codon usage profiles of 12 protein-coding genes (PCGs) and their nucleotide composition were calculated using Geneious 6.1.5 program (Biomatters Co., Auckland, New Zealand) [20]. For the analyses of transfer RNA genes, putative secondary structures of 22 tRNA genes were identified using the program tRNAscan-SE (http://lowelab.ucsc.edu/tRNAscan-SE), or by visual identification combined with manual proofreading through anticodon and tRNA secondary structures of trematodes which are available in GenBank.
Phylogenetic analysis
To assess the phylogenetic relationship of E. hortense with other digenean trematodes, the complete mt genome sequences of 24 trematode species were retrieved from GenBank and employed for phylogenetic analysis. The mtDNA sequences were as follows: Clinostomum complanatum (KM923964.1), Calicophoron microbothrioides (KR337555.1), Clonorchis sinensis (JF729303), Dicrocoelium chinensis (NC_025279), Dicrocoelium dendriticum (NC_025280), Fasciola gigantica (KF543342), F. hepatica (NC_002546), Haplorchis taichui (KF214770), H. conoideum (KM111525), Metagonimus yokogawai (KC330755), Ogmocotyle sikae (KR006934.1), Opisthorchis felineus (EU921260), O. viverrini (JF739555), P. cervi (KF475773), Paramphistomum leydeni (KP341657.1), Paragonimus westermani (NC_002354) Schistosoma haematobium (DQ157222), S. japonicum (NC_002544), S. mansoni (NC_002545), S. mekongi (NC_002529), S. spindale (DQ157223), S. turkestanicum (HQ283100), Trichobilharzia regenti (NC_009680), and using Monogenea species Gyrodactylus thymalli (NC_009682) as the outgroup. The nucleotide alignments involved PCGs were generated on the basis of the protein alignment using codon alignment amino acid sequences for 12 PCGs, which were individually aligned utilizing Clustal X 1.83 under default parameters [21]. Phylogenetic analyses were performed with the maximum parsimony (MP) method. Phylogenetic reconstructions were carried out using PAUP* Version 4.0b10 [22]. Phylograms were drawn using the Tree View program version 1.65 [23].
RESULTS
Features of the mt genome of E. hortense
The circular complete mt genome of E. hortense (GenBank accession no. KR062182) was 14,994 bp in length (Fig. 1). The mt genome encoded 36 genes: 12 PCGs (cox1-3, nad1-6, nad4L, atp6, cytb), 22 transfer RNA genes, and 2 ribosomal RNA genes (rrnL and rrnS). All genes were transcribed in the same direction. The relative positions and lengths of each gene are given in Table 2. The nucleotide contents in the mt genome were 41.77% (T), 21.26% (A), 24.79% (G), and 12.18% (C), and the overall A+T content of the mt genome was 63.03%.
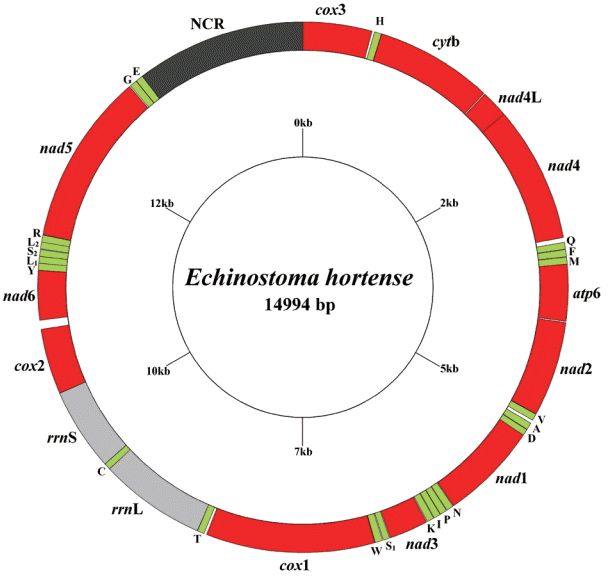
Arrangement of the mitochondrial genome for Echinostoma hortense. Gene scaling is approximate only. All genes have standard nomenclature, including the 22 tRNA genes, which are designated by the one-letter code for the corresponding amino acid, with numerals differentiating each of the 2 leucine and serine specifying tRNAs (L1 and L2 for codon families CUN and UUR, respectively; S1 and S2 for codon families AGC and UCA, respectively).

The organization of mitochondrial genome of Echinostoma hortense
A total of 3,349 amino acids were encoded by the E. hortense mt genome, and the length of PCGs was in the order: nad5>cox1>nad4>cytb>nad1>nad2>cox3>cox2>atp6>nad6>nad3>nad4L. The concatenated nucleotide sequences of 12 PCGs were 10,047 bp in length and composed of 45.13% T, 18.54% A, 24.73% G, and 11.60% C, with A+T accounting for 63.67% of the total nucleotides encoding the 12 PCGs. The initiation and termination codons of the above amino acid sequences were identified by sequence comparison with homologs in the mt genomes of other trematodes. The ATG codon was used in 8 PCGs (cox3, cytb, nad4, nad2, nad1, nad3, cox2, and nad5), and the GTG codon in 4r genes (nad4L, atp6, cox1, and nad5) as start codons. The TAG was used as stop codon in 5 genes (cytb, atp6, nad1, nad3, and cox2) and the TAA termination codon in the remaining 7 genes (cox13, nad4L, nad4, nad2, cox11, nad6, and nad5).
Total 22 tRNA genes, which ranged from 59 to 69 bp in length, were predicted from the mt genomes. Among the 22 tRNA genes, tRNA-SerAGC and tRNA-SerUCA have unpaired Darms which are replaced by the loops of 7-15 bp, but other 20 tRNA genes could be folded into the conventional cloverleaf structure (data not shown). The 2 ribosomal RNA genes were inferred, rrnL was located between tRNA-Thr and tRNA-Cys, and rrnS was located between tRNA-Cys and cox2. The rrnL and rrnS of E. hortense were 973 bp and 759 bp in length, respectively. The A+T contents of the rrnL and rrnS of E. hortense mt genome were 61.63% and 61.00%, respectively. Only 1 AT-rich non-coding region was inferred in the mt genome, and the AT-rich region located between nad5 and cox3, which is 1,553 bp long with A+T of 64.58%.
Phylogenetic analysis
Phylogenetic analysis of concatenated nucleotide sequence datasets for all 12 PCGs were performed using MP method, and the result showed that Opisthorchiidae, Heterophyidae, Paragonimidae, Dicrocoeliidae, Fasciolidae, Echinostomatidae, Paramphistomidae, Notocotylidae, and Clinostomidae formed 1 group, whereas Schistosomatidae formed another group (Fig. 2). In former group, besides the bird fluke Clinostomum complanatum, other trematodes gathered together and formed 1 clade. In this clade, Fasciolidae, Echinostomatidae, Paramphistomidae, Notocotylidae, and Clinostomidae trematodes belonged to order Echinostomida all formed monophyly. E. hortense and H. conoideum gathered together, and they were closer to each other than to the Fasciolidae and order Echinostomida trematodes with high nodal support (Fig. 2).
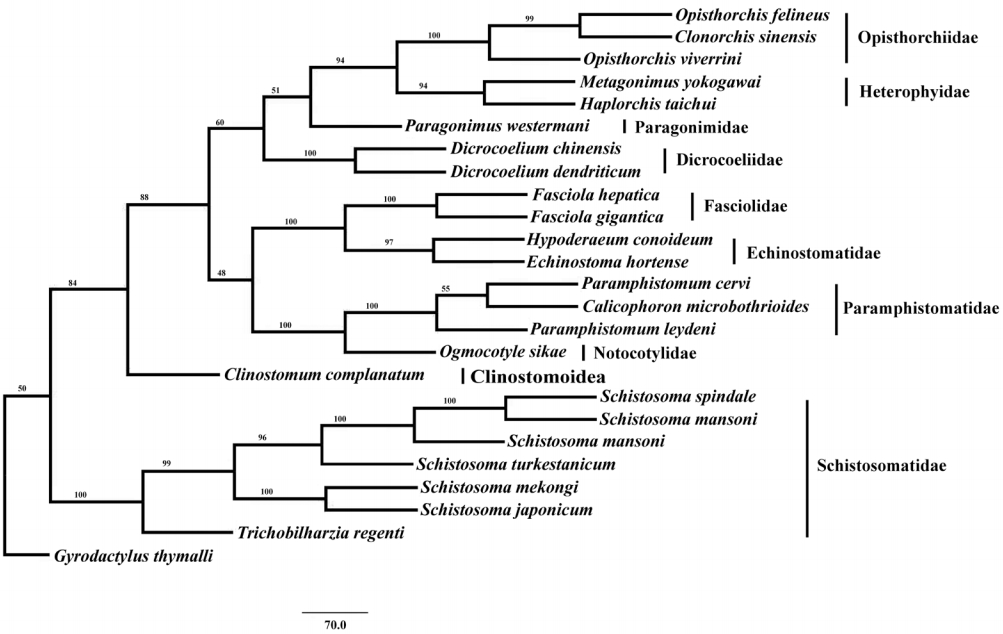
Phylogenetic relationship of Echinostoma hortense with other digenean trematodes based on mitochondrial genome sequence data. The concatenated amino acid sequences of 12 protein-coding genes were analyzed with maximum parsimony (MP) method, using Monogenean species Gyrodactylus thymalli (NC_009682) as the outgroup.
DISCUSSION
The complete mt genome of E. hortense (14,994 bp) is smaller than those of Eurytrema pancreaticum (15,031 bp), H. taichui (15,130 bp), M. yokogawai (15,258 bp), S. haematobium (15,003 bp), and S. spindale (16,901 bp), but slightly larger than those of other digenean species available in GenBank to date. The mt genome of E. hortense lacks atp8 gene, which is similar to the mt genomes of other trematodes [12,13,16,18]. All genes were transcribed in the same direction, which is consistent with the mt genomes of other digeneans [12,13,16,18,24]. The gene arrangement of 36 genes in E. hortense was the same as that of the H. conoideum (KM111525), P. cervi (KF475773), D. dendriticum (NC_025280), and D. chinensis (NC_025279), but different from that of other digenean species. Notably, there was a 28-64 bp long overlap between nad4L and nad4 in fluke mt genomes, but mostly was 28 bp or 40 bp in length. In E. hortense, nad4L and nad4 overlapped by 40 bp in size which is consistent with that of O. felineus, C. sinensis, F. hepatica, P. westermani, P. cervi, and H. taichui [13,16,18,24,25]. Though E. hortense and H. conoideum belong to the same family of Echinostomatidae, the lengthes of their cox3 genes were very different, with that of E. hortense shorter than H. conoideum by 312 bp.
The mt genome of E. hortense had 22 tRNA genes, like the majority of trematode mt genome sequences, except for that of P. westermani Korean isolate (NC_002354) and Indian isolate which have 23 and 24 tRNAs [24], respectively. The rrnL of E. hortense, at 973 bp, was the shortest among trematodes recorded yet, and the rrnS (759 bp) within the range of typical sizes for digenean mt genomes (700-800 bp). Usually, the size of non-coding region of trematode mt genome is between 55 bp and 2,500 bp, some of the trematode mt genomes have 2 non-coding regions (F. hepatica, F. gigantica, P. cervi, D. dendriticum, and D. chinensis), and a few flukes only contain 1 region (T. regenti, S. mekongi, H. conoideum, and H. taichui). Similarly, the mt genome of E. hortense only contains 1 non-coding region. In most trematode mt genomes, between genes or between tRNAs, there are many intergenic sequences with their lengths ranging usually from 1 bp to 50 bp, and the longest intergenic region in E. hortense mt genome reached 82 bp, but it was not AT-rich (45.12%) region.
There were many studies about phylogenetic status in the family Echinostomatidae have been done by some researchers. For example, Kudlai et al. [26] used 28S rDNA to study the phylogeny of 25 species of the superfamily Echinostomatoidea by Bayesian analysis. The results showed that 6 clades were clustered, and family Echinostomatidae was the largest group. Echinostoma and Echinoparyphium clustered together in a weakly-supported clade while the members of the other genera formed a 100% supported grouping. Echinostoma had a closer relationship with Echinoparyphium than to other genera of the family Echinostomatidae. In order to assess the relationships of Echinostoma with other closely related echinostomatids, 37 nad1 sequences and 15 ITS sequences of the genera Echinostoma, Echinoparyphium, Isthmiophora, and Hypoderaeum were analyzed by Kostadinova et al. [27]. The results showed that the genus Echinostoma is not a monophyletic group, and that E. hortense is most distantly related with other Echinostoma trematodes. This result was reconfirmed by Saijuntha et al. [28], who studied the phylogenetic relationships of Echinostoma and Hypoderaeum based on the cox1 sequences, and the results revealed that E. revolutum and E. malayanum of genus Echinostoma did not form a monophyletic clade and/or sister taxa. In contrast, a study of the phylogenetic relationships of North American echinostomes indicated that the genus Echinostoma is monophyletic, and the genera Hypoderaeum and Echinoparyphium are closely related as sister genera [29]. These contradictory results may be related to differences in geographical localities, sampled numbers, and the snail hosts of the echinostomes.
Therefore, Saijuntha et al. [28] suggested that the traditional morphological taxonomy of echinostomes, which is based on the size, shape, and arrangement of collar spines, the development of circumoral disc and also the testicular shape, should be reconsidered. The phylogenetic analysis in the present study indicated that the 2 fluke species, E. hortense and H. conoideum, of the family Echinostomatidae gathered together, which were closely related than to trematodes of the family Fasciolidae and other Echinostomida trematodes with high nodal support. This result was similar to the findings of previous reports. Although E. hortense and H. conoideum mt genomes were sequenced, more sequences of echinostomatid trematodes mt genomes are still needed to assess the phylogenetic relationships of these trematodes.
In conclusion, the present study determined the complete mt genome sequences of E. hortense, the first mt genome sequence of the genus Echinostoma of Echinostomatidae. The mtDNA dataset provided robust genetic evidence that E. hortense and H. conoideum were closer to each other than to the Fasciolidae and other Echinostomida trematodes, as well as provided new and useful genetic marker for further studies of the taxonomy, identification, and systematics of echinostomatid flukes.
Acknowledgements
This work was supported, in part, by the Fund for Imported Talents in Heilongjiang Bayi Agricultural University (XYB2014-07), and the Open Funds of the State Key Laboratory of Veterinary Etiological Biology, Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (grant no. SKLVEB2014KFKT006).
Notes
We have no conflict of interest related to this study.