Multiplex-Touchdown PCR to Simultaneously Detect Cryptosporidium parvum, Giardia lamblia, and Cyclospora cayetanensis, the Major Causes of Traveler’s Diarrhea
Article information
Abstract
This study aimed to develop a multiplex-touchdown PCR method to simultaneously detect 3 species of protozoan parasites, i.e., Cryptosporidium parvum, Giardia lamblia, and Cyclospora cayetanensis, the major causes of traveler’s diarrhea and are resistant to standard antimicrobial treatments. The target genes included the Cryptosporidium oocyst wall protein for C. parvum, Glutamate dehydrogenase for G. lamblia, and 18S ribosomal RNA (18S rRNA) for C. cayetanensis. The sizes of the amplified fragments were 555, 188, and 400 bps, respectively. The multiplex-touchdown PCR protocol using a primer mixture simultaneously detected protozoa in human stools, and the amplified gene was detected in >1×103 oocysts for C. parvum, >1×104 cysts for G. lamblia, and >1 copy of the 18S rRNA gene for C. cayetanensis. Taken together, our protocol convincingly demonstrated the ability to simultaneously detect C. parvum, G. lamblia, and C. cayetanenesis in stool samples.
INTRODUCTION
Traveler’s diarrhea (TD) is the most common and persistent travel-related disease [1,2]. TD pathogens are well known for causing an urgent loose stool, severe abdominal pain, vomiting, chills, and fever [1,3]. According to the CDC’s Health Information for International Travel 2016, the attack rates of TD ranged from 30% to 70% of travelers, depending on the destination and season [4]. In addition, the GeoSentinel database from which travel-related morbidity is recorded reported 335 diarrhea cases per 1,000 medical visits after traveling [5]. In general, enterotoxigenic Escherichia coli (ETEC), enteroaggregative E. coli (EAEC), Salmonella species, Shigella species, Aeromonas species, and Campylobacter jejuni are considered the major bacterial pathogens that can elicit TD [1]. Moreover, viruses causing gastroenteritis (e.g., noroviruses and rotavirus), as well as waterborne protozoan parasites (e.g., Cryptosporidium parvum, Giardia lamblia, Cyclospora cayetanensis, and Entamoeba histolytica), are also important TD pathogens [1]. In particular, waterborne protozoan parasites have been reported as the causes of endemic outbreaks in some countries [6]. Because the risk for cyclosporiasis has been increasing in some regions [1,7,8], C. parvum, G. lamblia, and C. cayetanensis have attracted attention as the cause of TD when travelers to developing countries cannot be cured by the standard antimicrobial treatments for their diarrhea [1].
However, there are few existing rapid diagnostic tools that can save time and effort by simultaneously detecting such protozoan parasites. This study aimed to develop a simultaneous multiplex PCR method for detecting 3 species of waterborne protozoan parasites (C. parvum, G. lamblia, and C. cayetanensis) causing TD accompanied by diarrhea that persists for longer than 2 weeks. For this purpose, we used the genomic DNA of these parasites, as well as the gene targets; Cryptosporidium oocyst wall protein (COWP) gene for C. parvum, the glutamate dehydrogenase (gdh) gene for G. lamblia, and the 18S ribosomal RNA (18S rRNA) gene for C. cayetanensis were selected by a preliminary in silico analysis. Here, we report the development of a successful protocol to avoid mispriming and non-specific binding among primer pairs using a touchdown PCR method [9]. In addition, we investigated the limit of detection (LOD) for a mixture of primers and optimized the PCR conditions for the detection of each parasite in protozoa-spiked human stool samples.
MATERIALS AND METHODS
DNA preparation
For the preparation of the genomic DNA, C. parvum oocysts were purchased from Waterborne™, Inc. (New Orleans, Louisiana, USA) and G. lamblia cysts were kindly provided by the Division of Malaria and Parasitic Diseases, Korea National Research Institute of Health, Korea Center for Disease Control and Prevention, Osong, Korea. C. cayetanenesis was purchased from ATCC as synthetic DNA, including the full 18S rRNA, internal transcribed spacer 1 (ITS1), and internal transcribed spacer 2 (ITS2) (ATCC® PRA-3000SD™, Manassas, Virginia, USA). Genomic DNA for 1×106 oocysts/cysts of C. parvum and G. lamblia were extracted using a DNeasy blood and tissue kit (Qiagen, Hilden, Germany) with a repeated freezing and thawing process (6 cycles of 95°C for 1 min and liquid nitrogen for 30 sec). Finally, the purified DNA was eluted by 20 μl of AE buffer using a mini spin column (Qiagen) and stored at −20°C. The purity and concentration of the DNA were measured using Nanodrop 2000 (Thermo Scientific, Wilmington, Delaware, USA).
DNA extraction from oocysts and the cyst-spiked human stool sample
Stool samples were obtained from health screening participants according to the Ethics Committee of the Inha University Hospital IRB approved protocol (Research 15-026) and provided for the preparation of the oocysts and cyst-spiked stool sample. C. parvum oocysts were decimally diluted in PBS to obtain a dilution series containing 1×107–1×103 and seeded into 200 mg of uninfected human stool samples. Protozoan parasites were not found in these stool samples using standard detection tests comprising microscopy and conventional nested PCR [10,11]. Likewise, human stool samples containing G. lamblia cysts were prepared using the method described above. Genomic DNA in the C. parvum or G. lamblia-spiked stool sample was extracted using the AccuPrep® Stool DNA extraction kit (Bioneer, Daejeon, Korea) according to the manufacturer’s protocol with a repeated freezing and thawing process (6 cycles at 95°C for 1 min and liquid nitrogen for 30 sec) to make it easier to break the cyst wall. Finally, the total genomic DNA was eluted by 20 μl of elution buffer (Bioneer), and 2 μl was used for the PCR reaction. In the case of C. cayetanensis, we used synthetic DNA purchased from ATCC because it was difficult to obtain the necessary amount of cysts required for this experiment. After 1×106 copies of synthetic DNA (ATCC) were serially diluted to 10−1 copies, 1 μl of the diluted DNA, which contains each copy number from 1×106 to 1×10−1, and 2 μl of the stool genomic DNA extracted from 200 mg of an uninfected stool sample were mixed and applied to the PCR reaction as a template. Although the copy number of the 18S rRNA in C. cayetanensis has not been elucidated, it can be predicted to be greater than 100 copies when it is compared to that of other protozoa with similar phylogenetic features [12,13].
Primer design
Diagnostic PCR primers were designed to amplify the specifically targeted gene segments for each parasite (Table 1). All primers were newly designed in the present study. Briefly, the primers for C. parvum were developed with some modifications based on previously reported primers, cry-9 and cry-15, targeted to the Cryptosporidium oocyst wall protein (COWP) [14]. In contrast, the primers for G. lamblia and C. cayetanensis were created using Geneious version R8 software (Biomatters Ltd., Auckland, New Zealand) for GDH and the 18S rRNA, respectively. Primer synthesis and sequencing for amplicon were performed by Macrogen Inc. (Seoul, Korea).

Information regarding the primers used for simultaneous multiplex-touchdown PCR of C. parvum, G. lamblia, and C. cayetanensis
Multiplex-touchdown PCR amplification
Multiplex-touchdown PCR was performed using Surecycler 8800 (Agilent Technologies, Santa Clara, California, USA), and the PCR conditions were performed as follows. The DNA templates (1–3 μl), 15 μl of 2×PCR premix (2×Taq PCR Pre-Mix, Solgent Co., Daejeon, Korea), and 5 μl of the primer mixture (consisting of 10 pmol primers in each parasite) was mixed with HPLC-grade distilled water (Wako Pure Chemical, Osaka, Japan) to a total volume of 30 μl. The PCR reaction for the negative control was reacted with only the primer, without protozoan DNA. At this time, the specificity and sensitivity of each primer were previously tested and confirmed using a single PCR amplification (data not shown). The multiplex-touchdown PCR amplification protocol consisted of 5 min at 95°C for pre-denaturation, 20 cycles of denaturing at 95°C for 30 sec, annealing at 65°C (with 0.2°C decrements from 65°C to 61.2°C at every cycle) for 40 sec, and extension at 72°C for 1 min. This was followed by a further 25 cycles of denaturing at 95°C for 30 sec, annealing at 61.2°C for 40 sec, and extension at 72°C for 1 min. The reaction was finished with a final extension for 5 min at 72°C. The PCR products were confirmed by the StaySafe Nucleic Acid Gel Stain (Real Biotech Corporation, Taipei, Taiwan) and photographed with UV transillumination using the DNR MiniLumi (DNR Bio-Imaging Systems, Jerusalem, Israel) gel documentation system after loading on 2% agarose (Duchefa, Haarlem, The Netherlands)-TAE gels.
RESULTS
Target specificity of primers and multiplex-touchdown PCR protocol
The amplicon size for each parasite was 555 bp for C. parvum, 188 bp for G. lamblia, and 400 bp for C. cayetanensis (Fig. 1). The single PCR products were confirmed by analyzing the DNA sequences. Each amplicon exhibited a 100% similarity to AB089292 (C. parvum), KJ499992 (G. lamblia), and AB111183 (C. cayetanensis) in the GenBank database (http://www.ncbi.nlm.nih.gov) (data not shown). In every reaction, the primers (a primer mixture containing all 3 types of primers) did not show any cross-reactivity in the parasite-free stools or other protozoan parasites (i.e., E. histolytica, Toxoplasma gondii, and Babesia microti), which was previously screened for specificity (data not shown). In Fig. 1, the primer mixture was reacted with each target for monoplex PCR (lanes 2–4), 2 types of targets for duplex PCR (lanes 5–7), and all 3 kinds of targets for triplex PCR (lane 8). The amount of template DNA used for each PCR reaction corresponds to 2 μl of the 20 μl total DNA solution extracted from 1×106 oocysts and cysts of C. parvum or G. lamblia, and 1 μl of the C. cayetanensis synthetic DNA (equivalent to 103 copies). Our results show that the multiplex PCR protocol using the touchdown PCR method did not result in cross-reactivity between the primers or template DNA in the mixed condition of primers and template DNA. In addition, we suggest that the PCR protocol developed in this study makes it possible to improve the specificity of PCR amplification (Fig. 1).
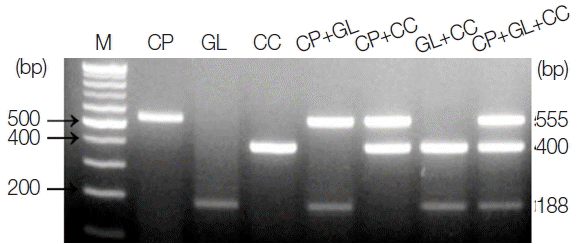
PCR products amplified using a primer mixture (CP-MT-F, CP-MT-R, GL-MT-F, GL-MT-R, CC-MT-F, and CC-MT-R). Lane 1, 100-bp marker; lanes 2 to 4, single PCR products [C. parvum (555 bp, lane 2), G. lamblia (188 bp, lane 3), and C. cayetanensis (400 bp, lane 4)]; lanes 5 to 7, duplex PCR products [C. parvum+G. lamblia (lane 5), C. parvum+C. cayetanensis (lane 6)], and G. lamblia+C. cayetanensis (lane 7)]; Lane 8, triplex PCR (C. parvum+G. lamblia+C. cayetanensis).
Limit of detection of each parasite in parasite-spiked human stool samples
To investigate the LOD of these protozoa for clinical diagnosis, C. parvum- and G. lamblia-spiked stool samples (which include between 1×107 and 1×103 oocysts and cysts, respectively) were prepared, and their genomic DNA were used as a template for further PCR reactions. Moreover, 1 μl of synthetic C. cayetanensis DNA, which was diluted from 1×106 to 1×10−1 copies, and 1 μl of the uninfected stool genomic DNA (100 ng/μl) were mixed and used as a template for the PCR reaction. To investigate the specificity and cross-reactivity of the primers, a primer mixture was used in every reaction (Fig. 2A–C). Our results indicated that the LOD of C. parvum was higher than 1×103 oocysts (Fig. 2A), and the LOD of G. lamblia was greater than 1×104 cysts (Fig. 2B). Additionally, the LOD of C. cayetanensis was higher than 1×100 copies in the stool DNA (Fig. 2C). This result suggests that the primer consisting of a ribosomal DNA with a high copy number developed for C. cayetanensis is highly sensitive compared to that of C. parvum and G. lamblia, considering the differences in the species of protozoa and the characteristics of the target DNA.
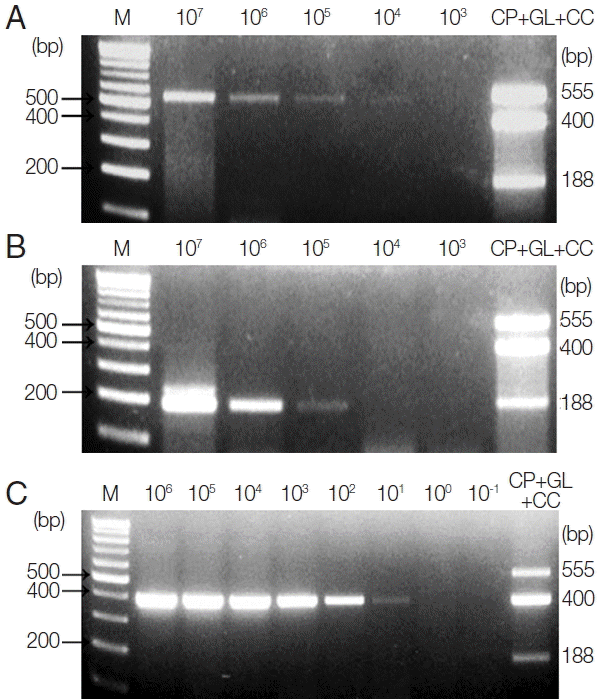
Limit of Detection (LOD) of each parasite in parasite-spiked human stool samples. To investigate the LOD of each parasite when the primer mixture is applied to the human stool samples, C. parvum, G. lamblia, and C. cayetanensis-spiked human stool samples were prepared as described in the “Materials and Methods” section. (A) Lane 1, 100 bp marker; lanes 2 to 6, stool DNA samples containing serially diluted (107 to 103) C. parvum oocysts; lane 7, the results of triplex PCR (a mixture of the target DNA). (B) Lane 1, 100 bp marker; lanes 2 to 6, stool DNA samples containing 107 to 103 serially diluted G. lamblia cysts; lane 7, the results of the triplex PCR (a mixture of target DNAs). (C) lane 1, 100 bp marker; lanes 2 to 9, Cyclospora-negative stool DNA samples mixed with serially diluted quantitative synthetic Cyclospora cayetanensis DNA (ATCC, PRA-3000SD™) in copy numbers ranging from 106 to 10−1; lane 10, the result of triplex PCR (a mixture of target DNAs). The LOD was investigated by a common multiplex-touchdown PCR protocol using a primer mixture.
DISCUSSION
TD is a major public health concern that can result in significant morbidity and disability [2]. A common cause of TD is enteric bacterial infections; however, waterborne protozoa are also a globally important cause of TD because they are easily transmitted through water and food [1,14,15]. Furthermore, it is important to develop a conventional and rapid detection method for clinical applications because TD caused by C. parvum, G. lamblia, and C. cayetanensis are resistant to standard antimicrobial treatment [1,16]. Given these issues, this study aimed to develop a new specific multiplex PCR method applicable to fecal samples.
There are several different methods to detect waterborne protozoa, including microscopic examination, PCR-based techniques, fluorescence in situ hybridization, and immunology-based methods [17]. Among them, PCR is the most common method for molecular detection, and its results are more sensitive than microscopic observations in detecting the parasite [18]. In particular, the multiplex PCR method has been developed for targeting enteric protozoa (e.g., C. parvum, G. lamblia, and E. histolytica) [19–21]. However, Cyclospora infections have recently been increasing in association with poverty and areas of poor sanitation, with fecal-contaminated soil creating an important transmission vehicle for Cyclospora [6,22]. C. cayetanensis infection in patients with TD has been reported in individuals who live in, or have visited different areas of the United States, Caribbean Islands, Central America, South Asia, Eastern Europe, Morocco, and Nepal [23]. In this situation, the laboratory diagnosis of the infection must be followed for the rapid resolution of symptoms. Similar to C. parvum and G. lamblia, a reliable detection method for C. cayetanensis was developed using a specific PCR assay [24]. However, we have undertaken the additional challenge of effectively identifying multiple outbreaks of water and foodborne protozoa infections by developing a simultaneous multiplex detection method. For this purpose, we designed a novel multiplex PCR method for the simultaneous detection of C. parvum, G. lamblia, and C. cayetanenesis as potent pathogenic protozoa causing TD. Firstly, we tested several target genes of these protozoa via an in silico analysis using Geneious software to ensure the specific amplification and avoidance of cross-reactivity with other protozoan parasites or enteric bacterial pathogens. As the result, COWP, gdh, and 18S rRNA were selected for C. parvum, G. lamblia, and C. cayetanenesis, respectively. These genes are commonly used for classifying species and identifying parasite isolates using molecular analyses [13,25,26]. The specificity of the primers developed in the present study was demonstrated by the specific amplification of single, duplex, and triplex PCR. Furthermore, the amplified gene fragments exhibited a 100% identity in comparison with the query sequences in BLAST. In particular, the protocol adopted herein consists of touchdown PCR, which circumvents spurious priming during gene amplification [8], and thus, there is no non-specific amplification of any other microbial DNA in the stool samples.
In this study, the LOD for PCR amplification was more than 1×103 oocysts in C. parvum and more than 1×104 cysts in G. lamblia in 200 mg of the stool samples required by a routine Stool DNA extraction kit. The detectable number of oocysts from symptomatic individuals has been reported to be 1×105–1×107 oocysts per gram of feces in C. parvum [27], and the infection level may be detectable using our PCR method. In contrast, the LOD to G. lamblia was higher than the results obtained using a multicopy rRNA gene, in which the confirmation in the agarose gel was observed for a minimum of 2,000 trophozoites [28]. The problem in LOD using the gdh gene but not the multicopy rRNA gene can be overcome by the characteristics of the gdh gene, which is more sensitive and specific than the triose phosphate isomerase gene used prevalently for G. lamblia detection [29]. Above all, our choice to use the gdh gene is related to the use of the 18S rRNA gene, a multicopy rRNA, for C. cayetanenesis to induce the difference in the amplicon size for the purpose of multiplex PCR development. In general, the PCR target for C. cayetanenesis was primarily an rRNA gene, such as ITS or 18S [25,30]. In addition, our preliminary data revealed that the C. cayetanenesis 18s rRNA was adequate for the multiplex PCR design because of the size difference and specificity in relation to C. parvum and G. lamblia (data not shown). The LOD of C. cayetanenesis was determined for the lowest amount (≥10 copies) of DNA that could be amplified using this system.
Taken together, our protocol has convincingly demonstrated the ability to simultaneously diagnose multiple protozoa in stool samples. Although future studies are required to further evaluate the sensitivity of using clinical samples, ours is the first attempted PCR-based method for the simultaneous detection of C. parvum, G. lamblia, and C. cayetanensis.
ACKNOWLEDGMENT
This study was supported by a grant from Korea Centers for Disease Control and Prevention, Republic of Korea (2015-E54003-00).
Notes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.