Genetic Diversity and Phylogenetic Analysis of the Iranian Leishmania Parasites Based on HSP70 Gene PCR-RFLP and Sequence Analysis
Article information
Abstract
Despite the broad distribution of leishmaniasis among Iranians and animals across the country, little is known about the genetic characteristics of the causative agents. Applying both HSP70 PCR-RFLP and sequence analyses, this study aimed to evaluate the genetic diversity and phylogenetic relationships among Leishmania spp. isolated from Iranian endemic foci and available reference strains. A total of 36 Leishmania isolates from almost all districts across the country were genetically analyzed for the HSP70 gene using both PCR-RFLP and sequence analysis. The original HSP70 gene sequences were aligned along with homologous Leishmania sequences retrieved from NCBI, and subjected to the phylogenetic analysis. Basic parameters of genetic diversity were also estimated. The HSP70 PCR-RFLP presented 3 different electrophoretic patterns, with no further intraspecific variation, corresponding to 3 Leishmania species available in the country, L. tropica, L. major, and L. infantum. Phylogenetic analyses presented 5 major clades, corresponding to 5 species complexes. Iranian lineages, including L. major, L. tropica, and L. infantum, were distributed among 3 complexes L. major, L. tropica, and L. donovani. However, within the L. major and L. donovani species complexes, the HSP70 phylogeny was not able to distinguish clearly between the L. major and L. turanica isolates, and between the L. infantum, L. donovani, and L. chagasi isolates, respectively. Our results indicated that both HSP70 PCR-RFLP and sequence analyses are medically applicable tools for identification of Leishmania species in Iranian patients. However, the reduced genetic diversity of the target gene makes it inevitable that its phylogeny only resolves the major groups, namely, the species complexes.
INTRODUCTION
Leishmaniasis, with diverse clinical manifestations, is a health problem in many countries, including Iran. There are 2 types of leishmaniasis in the country, cutaneous leishmaniasis (CL) and visceral leishmaniasis (VL). CL consists of 2 epidemiological forms, zoonotic CL caused by Leishmani major and anthroponotic CL caused by Leishmania tropica [1]. They are prevalent throughout the country, with tens of thousands of cases every year. In recent years, due to new settlements, population increase, urbanization, migration, and increase in sandfly populations, they have emerged in some new districts [2]. VL is caused by Leishmania infantum and has been reported sporadically throughout Iran; however, it is more prevalent in northwestern and southern parts of the country with approximately 100 to 300 reported cases annually [3,4].
The exact identification of Leishmania species is of great importance with concern to diagnostic, disease prognosis, epidemiological surveillance, and clinical studies. Many diagnostic techniques, including microscopy, in vitro culture, biochemical, immunological, and molecular approaches have been applied for detection and identification of Leishmania parasites. Clinical features and microscopic examinations are not suited for species identification of parasites [5]. Multi-locus enzyme electrophoresis (MLEE) has been known as the reference method for identification of Leishmania spp. [6]. However, this technique requires mass cultivation of parasites, and it lacks of power to differentiate the parasites below the species level [7,8]. On the other hand, PCR-based methods, including PCR-RFLP and DNA sequence analyses are highly informative [9]. RFLP technique, has allowed direct and rapid identification of Leishmania species [10–12]. For phylogenetic reconstruction, sequence analysis is preferred over RFLP or fingerprinting patterns [5]. During the past 2 decades, various DNA sequences have been used as targets to reconstruct the phylogeny within the genus Leishmania, including genes encoding the catalytic polypeptide of DNA polymerase a (polA) [13], the largest subunit of RNA polymerase II (rpoIILS) [13], 7SL RNA [14], ribosomal internal transcribed spacer (ITS) [15–17], the N-acetylglucosamine-1-phosphate transferase (NAGT) gene [18], the mitochondrial cytochrome b gene (cytb) [19], and recently, sequences of the heat-shock protein 70 gene (HSP70) subfamily [20].
The HSP70 is conserved across prokaryotes and eukaryotes, and the protein as well as its encoding gene have been applied in phylogenetic studies of many parasites [5], including Leishmania [20,21]. Despite the broad distribution of leishmaniasis among Iranians, little is known about the genetic characteristics of the causative agents across the country [22–24]. Plus, most of the studies, targeted the HSP70 gene sequences to date, have covered only small geographical areas, with limited number of isolates [22,24]. Applying both HSP70 PCR-RFLP and DNA, the present study was designed to evaluate the genetic diversity and phylogenetic relationships among Leishmania spp. isolated from Iranian patients across the country and available reference strains.
MATERIALS AND METHODS
Parasite strains and DNA isolation
Samples positive for leishmaniasis were taken from 24 Iranian (from almost all districts of Iran) and 3 Afghan patients (who lived in Iran) referred to either Leishmaniasis Laboratory at the School of Public Health or Center for Research and Training in Skin Diseases and Leprosy, Tehran University of Medical Sciences. Four rodents and 5 canine isolates were also included in the sample (Table 1; Fig. 1). For DNA isolation, samples were transferred to RPMI-1640 culture medium (Gibco, Frankfurt, Germany) supplemented with 10–15% fetal bovine serum (Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) and incubated at 24–26°C. Leishmania promastigotes were harvested from RPMI cultures and washed twice by PBS buffer and once by sterile distilled water. The pellets were kept frozen at −20°C until use. Genomic DNA was extracted from isolated parasites using the QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.

Details of the original and GenBank Leishmania isolates used in the present study

PCR amplification of HSP70 gene
Amplification was performed with 2 specific primers, HSP70sen (5′-GACGGTGCCTGCCTACTTCAAG-3′) and HSP70ant (5′-CCGCCCATGCTCTGGTACATCC-3′), previously used by Garcıa et al. [21]. They were synthesized by the Bioneer Corporation (Daejeon, South Korea). The PCR reaction mix (50 ml) contained 20 μl premix (Roche, Mannheim, Germany), 4 μl forward and reverse primers (10 pmol), 3 μl DNA (50–100 ng) template, and 20 μl double distilled water. The thermal cycling conditions consisted of initial denaturation at 94°C for 5 min, 30 cycles of 94°C for 5 min, 61°C for 1 min, and 72°C for 1 min 30 sec; and a final extension of 5 min at 72°C. Five ml of each PCR amplicon was run on a 1.2% agarose gel (Invitrogen, Carlsbad, California, USA), stained with ethidium bromide and visualized under UV light.
In silico and RFLP analysis of HSP70
The HSP70 gene PCR products (10 ml each) were digested with the HaeIII (BsuR1) restriction enzyme using the conditions recommended by the manufacturer (MBI Fermentas, Vilnius, Lithuania). At first, this enzyme was tested against sequences of reference strains (Fasta format genes downloaded from GenBank) and searched for restriction sites using Restriction Mapper 3 (www.restrictionmapper.org). Theoretically, obtained fragments from digestion with HaeIII would be expected to create different patterns as follows: L. major (351, 307, 246, 152, 99, 47, 41, 40, 34, and 2 bp), L. tropica (354, 338, 246, 150, 99, 80, 41, 40, 21, 13, and 8 bp), and L. infantum (338, 307, 246, 152, 99, 80, 53, 47, 41, 40, and 13 bp). Digestion was performed in a total of 30 ml 1× optimal buffer, using 1U HaeIII restriction enzyme. Reactions were incubated at 37°C and completely analyzed by electrophoresis in a 3% small fragment agarose gel. The gels were subsequently ethidium bromide stained and subjected to electrophoresis along with the Gene Ruler TM 50 bp DNA Ladder (MBI Fermentas) as a reference DNA size marker.
DNA typing and sequence analysis
PCR products of the HAP70 gene related to 36 Leishmania isolates were purified and sequenced using an ABI PrismTM 3730 Genetic Analyzer (Applied Biosystems, Foster City, California, USA) by the Macrogen Company (Seoul, South Korea). Sequencing was carried out by the same primer pair. Consensus sequences from forward and reverse reads were trimmed and edited using the BioEdit 5.0.9 [25]. Furthermore, HSP70 sequences of 37 reference strains available in GenBank database were taken from NCBI (http://www.ncbi.nlm.nih.gov) and included in the analyses. All the original and retrieved sequences were aligned together, first applying the ClustalW algorithm implemented in the MEGA 6.0 [26], and then manually. Applying the corrected Akaike Information Criterion (AICc) [27] in jModelTest 2.1.10 [28], the best-fit models of nucleotide substitution was estimated. The maximum likelihood (ML) and neighbor joining (NJ) phylogenetic trees were constructed using the MEGA software. The HSP70 gene sequence of Trypanosoma rangeli (GenBank accession No. EF108422) was used as an outgroup. Using Arlequin 3.11 [29], basic parameters of genetic diversity were estimated. Using the same software, demographic histories were examined based on 2 neutrality tests, Tajima’s D [30] and Fu’s Fs [31].
RESULTS
RFLP analysis
PCR amplification of the HSP70 gene resulted in production of about 1,420 bp fragment in all 36 Leishmania isolates. The amplicons of 20 isolates were subjected to PCR-RFLP analysis. Banding patterns were created with accordance to the expected profiles that were theoretically obtained from restriction map. The results of HSP70 PCR-RFLP presented 3 different electrophoretic patterns (Fig. 2), each identical to that of 1 of the 3 Leishmania species endemic in Iran, L. tropica (35%), L. major (50%), and L. infantum (15%). No intraspecific variation within the isolates of each species was observed.

Agarose gel (3%) showing 3 HSP70 PCR-RFLP profiles for Leishmania reference strains after digestion with HaeIII. Lane1, L. tropica; lane 2, L. tropica; lane 3, L. major; lane 4, L. major, lane 5, L. infantum; lane 6, L. infantum; lane M, 100-bp size marker.
Phylogenetic analyses
The best-fit models of nucleotide substitution, GTR+I+G, was applied to reconstruct phylogenetic relationships among all HSP70 sequences based on ML and NJ methods. The ML analysis (Fig. 3) grouped all the HSP70 sequences into 2 major clades, L. (Leishmania) clade comprising all Old World species and L. mexicana complex from the New World, and L. (Viannia) consisting of only New World species. The NJ tree (not shown) had similar subdivision. Phylogenetic analysis of the Iranian HSP70 gene sequences yielded a phylogenetic grouping in complete conformity with the primary PCR-RFLP identification. Iranian lineages, including L. major, L. tropica, and L. infantum were distributed among 3 complexes L. major, L. tropica, and L. donovani. However, within the L. major and L. donovani complexes, the HSP70 phylogeny was not able to distinguish clearly between the L. major and L. turanica and between the L. infantum, L. donovani, and L. chagasi isolates, respectively. Furthermore, no evidence for phylogeographic structure and substructure was evident within the phylogenetic tree.

ML tree summarizing the relationship between HSP70 sequences of Leishmania species. Bootstrap resampling values are provided at each fork (given only if ≥40%). Iranian isolates are represented by circle (L. major), triangle (L. tropica), and square (L. infantum) symbols.
Genetic diversity and demographic history
The sequence analysis of 1,167 bp fragment of the HSP70 gene among 36 Leishmania detected 12 polymorphic sites (9 singletons and 12 parsimony informative sites) among sequences (Table 2). Genetic diversity estimates of 3 Leishmania species are summarized in Table 2. In total, 6 haplotypes were identified, 3 in L. major, 2 in L. tropica, and 1 in L. infantum. Haplotype diversity (h) was ranging from 0.53±0.136 for L. tropica to 0 for L. infantum. Nucleotide diversity was ranging from 0.436±0.13 for L. tropica to 0 for L. infantum. Neutrality tests presented different results, including significant and negative Tajima’s D and non-significant Fu’s Fs values for L. major, and non-significant and positive Tajima’s D and Fu’s Fs values for L. tropica. Accordingly, these results provided support for rejecting the hypothesis of population growth in each species.
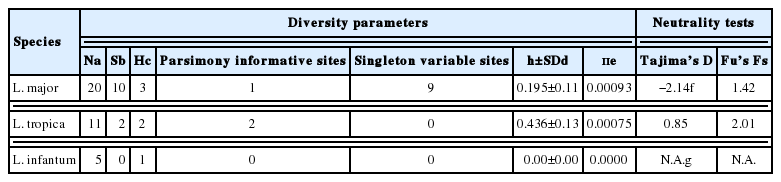
Basic parameters of genetic diversity and neutrality tests, for 3 Iranian Leishmania species
DISCUSSION
To date, most of the studies, targeting the HSP70 gene sequence in Iran, have covered only small geographical areas, with limited number of samples [22,24]. In the present study, using both PCR-RFLP and sequence analysis methods, we have extended previous observations by analyzing the HSP70 gene sequence in a larger cohort consisting of samples from almost all districts across the country.
Totally, 36 human and animal cases, including 27 CL, 5 VL, and 4 rodents were examined. Application of PCR-RFLP method on HSP70 gene clearly presented 3 different electrophoretic patterns, each belonged to 1 of the 3 Leishmania species in Iran. From 27 culture-positive samples prepared from CL cases, 15 (55.5%) were infected with L. major and 12 (44.5%) with L. tropica. All 5 canine specimens were infected with L. infantum, and all 4 rodents were infected with L. major. These results are in agreement with the results from the previous study on Iranians by Hajjaran et al. [23]. In their study, from 112 culture-positive samples prepared from CL cases, 75 (67%) were infected with L. major and 37 (33%) with L. tropica; from 25 rodents, 21 were infected with L. major; and from 28 culture-positive samples prepared from VL cases, 26 were infected with L. infantum.
Genus Leishmania is considerable for a large number of described species. They have been described as distinct species mainly according to biological, clinical, geographical, immunological, epidemiological, and biochemical measurements [5]. Though numerous molecular techniques and markers have been proposed for resolving the taxonomy of Leishmania [13–20], describing a Leishmania species or embracing all of the described taxa is still not straightforward. The MLEE method have been considered as the reference technique by many authors [6]. Application of this technique resulted in the present Leishmania classification system, comprising 17 described species [8,32–34]. Based on this classification, the genus Leishmania consists of the 2 subgenera, L. (Leishmania) comprising all Old World species and L. mexicana complex from the New World, and L. (Viannia) consisting of only New World species [5]. Analysis of several DNA sequences as targets during the past 2 decades [13–20] have been consistent in that each of the 2 subgenera formed a distinct monophyletic clade and that a deep phylogenetic gap within L. (Leishmania) separated species of the Old and New World. Furthermore, in all of these studies, L. (Viannia) was closest to the root, while L. (Leishmania) formed the crown of the trees, an indication of New World origin of Leishmania [5,6].
Phylogenetic analysis of the genus Leishmania based on the HSP70 gene sequence by Fraga et al. [20] which also supported the above grouping, has included by far the largest number of species. Interestingly, only 8 monophyletic clades were deciphered in their phylogenetic trees and networks, 4 in L. (Leishmania) and 4 in L. (Viannia). Accordingly, the authors concluded that the HSP70 phylogeny supports only 8 species, namely, L. donovani, L. major, L. tropica, L. mexicana within L. (Leishmania) and L. lainsoni, L. guyanensis, L. naiffi and L. braziliensis within L. (Viannia), and that the concept of species complexes should be abandoned. Subsequently, inspired by these results, some authors voted for revising the taxonomy of the genus Leishmania [6,35].
In our study, application of PCR-RFLP method on HSP70 gene clearly presented 3 different electrophoretic patterns, each belonged to 1 of the 3 Leishmania species in Iran. However, PCR-RFLP method was not able to detect any intraspecific variations. The efficiency of this method for identification of Leishmania species has already been demonstrated by some authors [21,23,24,36]. On the other hand, analysis of HSP70 DNA sequences of Leishmania isolates was more informative. Despite the low levels of genetic diversity among Iranian isolates, sequence analysis was able to identify 6 haplotypes, 3 belonged to L. major, 2 belonged to L. tropica, and 1 haplotype belonged to L. infantum. Within L. (Leishmania), Iranian lineages, including L. major, L. tropica, and L. infantum, were distributed among 3 species complexes L. major, L. tropica, and L. donovani. However, within both L. major and L. donovani complexes, the HSP70 phylogeny could not show clear discrimination between previously described species. HSP70 is under stabilizing selective pressure for conservation of function since synonymous substitutions (6.3%) are favored over non-synonymous (1.9%) [20]. The reduced genetic diversity of the HSP70, makes it inevitable that its phylogeny only resolves the species complexes and prohibits valid deductions regarding the species and subspecies levels [35].
Overall, given that only a limited number of Leishmania species causes leishmaniasis among Iranians (namely, L. major, L. tropica, and L. infantum), our results indicated that both HSP70 PCR-RFLP and DNA sequence analysis are medically applicable tools for identification of Leishmania species in Iranian patients. However, in agreement with previous authors [6,20,35], our results support the idea of revising the taxonomy of the genus Leishmania. Otherwise, in the future assessments, species assignment in Leishmania should be according to highly variable DNA sequences with considerable congruency [35]. For resolving taxonomic ambiguities within the major groups, including Leishmania, highly discriminatory methods such as multi-locus sequence typing (MLST) and genome-wide single nucleotide polymorphisms (SNPs) are probably better suited.
ACKNOWLEDGMENTS
The authors would like to thank the staff of the School of Public Health and Center for Research and Training in Skin Diseases and Leprosy, Tehran University of Medical Sciences. This research was supported by a grant from Zanjan University of Medical Sciences, and was a part of the first author’s doctoral dissertation.
Notes
CONFLICT OF INTEREST
We have no conflict of interest related to this study.