Suppressors for Human Epidermal Growth Factor Receptor 2/4 (HER2/4): A New Family of Anti-Toxoplasmic Agents in ARPE-19 Cells
Article information
Abstract
The effects of tyrosine kinase inhibitors (TKIs) were evaluated on growth inhibition of intracellular Toxoplasma gondii in host ARPE-19 cells. The number of tachyzoites per parasitophorous vacuolar membrane (PVM) was counted after treatment with TKIs. T. gondii protein expression was assessed by western blot. Immunofluorescence assay was performed using Programmed Cell Death 4 (PDCD4) and T. gondii GRA3 antibodies. The TKIs were divided into 3 groups; non-epidermal growth factor receptor (non-EGFR), anti-human EGFR 2 (anti-HER2), and anti-HER2/4 TKIs, respectively. Group I TKIs (nintedanib, AZD9291, and sunitinib) were unable to inhibit proliferation without destroying host cells. Group II TKIs (lapatinib, gefitinib, erlotinib, and AG1478) inhibited proliferation up to 98% equivalent to control pyrimethamine (5 μM) at 20 μM and higher, without affecting host cells. Group III TKIs (neratinib, dacomitinib, afatinib, and pelitinib) inhibited proliferation up to 98% equivalent to pyrimethamine at 1–5 μM, but host cells were destroyed at 10–20 μM. In Group I, TgHSP90 and SAG1 inhibitions were weak, and GRA3 expression was moderately inhibited. In Group II, TgHSP90 and SAG1 expressions seemed to be slightly enhanced, while GRA3 showed none to mild inhibition; however, AG1478 inhibited all proteins moderately. Protein expression was blocked in Group III, comparable to pyrimethamine. PDCD4 and GRA3 were well localized inside the nuclei in Group I, mildly disrupted in Group II, and were completely disrupted in Group III. This study suggests the possibility of a vital T. gondii TK having potential HER2/4 properties, thus anti-HER2/4 TKIs may inhibit intracellular parasite proliferation with minimal adverse effects on host cells.
INTRODUCTION
Toxoplasma gondii is an apicomplexan protozoa that is a ubiquitous obligate intracellular parasite. It is a zoonotic pathogen widespread in nature, in which felids are the definitive hosts, and all other warms blooded animals including humans can serve as intermediate hosts. Approximately 1/3 of humans worldwide are known to be chronically infected with T. gondii [1].
Almost all acquired infections are benign and transform into a chronic status especially in the central nervous system, but severe symptoms such as stillbirth, abortion or severe neurological disorders after delivery in congenital infection are also observed. These sometimes reactivate in immune compromised patients to cause toxoplasmic lymphadenitis, meningoencephalitis or ocular toxoplasmosis. Toxoplasmic retinochoroiditis is known to be the most common cause of infective posterior uveitis, and one of the major causes of visual impairment in highly endemic regions [2]. Antibiotics can reduce the number of recurrences and facilitate the resolution of inflammation in toxoplasmic retinochoroiditis, but a consensus on the utility of antibiotics has not been reached [3].
T. gondii creates a parasitophorous vacuole (PV) inside in which it develops further. ROP2 family of rhoptry proteins (ROPs) has a very important role in creating the parasitophorous vacuole membrane (PVM) within the host cells during this process. Some of these ROPs, and especially ROP16, have kinase domains in their C-terminal halves, which may function in signal transduction across the PVM as a protein kinase (PK) to maintain the host cell-parasite relationship and may be candidate targets for new drugs [4].
The majority of cellular pathways and especially those involved in signal transduction are regulated by PKs [5]. As one subgroup of PKs, protein tyrosine kinases (TK) are responsible for the activation of many proteins by phosphorylation that results from the binding of polypeptide ligands to cell surface receptors that possess tyrosine kinase catalytic activity. Phosphorylation of tyrosine residues result in downstream signal cascades. TKs can be classified into the receptor TKs (RTK) and the non-receptor TKs (NRTK) [6]. RTK family such as epidermal growth factor (EGF), fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF), and nerve growth factor (NGF) transduce extra-cellular signals to the cytoplasm by phosphorylating tyrosine residues on the receptors themselves (autophosphorylation) and on downstream signaling proteins. They are responsible for numerous signaling pathways within cells that lead to cell proliferation, differentiation, migration, or metabolic changes [6]. The large NRTK family, which includes Src, the Janus kinases (Jaks), and Abl, are integral components of the signaling cascades triggered by RTKs and by other cell surface receptors such as G protein-coupled receptors and receptors of the immune system. Numerous TKs have been identified as oncogenes in various tumors, so a strict regulation of their catalytic activity is an absolute requirement. They have also been implicated in various diseases such as diabetic retinopathy, atherosclerosis, psoriasis [7], and infections [4].
The importance of TKs in the survival of T. gondii in a hostile environment has been reported in a study by Muniz-Feliciano et al., who have reported the role of activation of EGFR in the blocking of autophagy protein-mediated killing of the parasite [8]. Peixoto et al. have shown by genomic analysis that T. gondii encodes 108 PK genes that are likely to have a catalytic activity, and 51 pseudokinases genes that lack a catalytic domain. Although most of these kinases can be classified into one of the major kinase groups, 78 of the 108 PKs lack an obvious ortholog in humans or yeast. These PKs play crucial roles in the proliferation and differentiation of the parasite. In addition, 55% are species specific while 15% are secretory, such as the rhoptry kinases mentioned above [9]. These differences of T. gondii PKs in structure and function from mammalian PKs can be used as drug targets. The functions of some of these PKs have been revealed and their inhibitors have been found, which look promising as novel drugs against toxoplasmosis [4,10–15].
PKs, and also T. gondii PKs, can be inhibited by so called ‘small molecule kinase inhibitors’ which have been developed for the chemotherapy of various types of cancer [16]. A tyrosine kinase inhibitor (TKI) is such a pharmaceutical drug that inhibits TKs [4], and numerous TKIs developed recently for the chemotherapy of cancers have expanded their efficacy for the treatment of infectious diseases [17,18]. In the present study we evaluated the effects of TKIs on the growth inhibition of intracellular T. gondii inside ARPE-19 cells, and tried to determine their mechanisms.
MATERIALS AND METHODS
Ethics statement
All procedures and handling of mice were conducted under an approved protocol by the Institutional Animal Care and Use Committee (IACUC) at the School of Medicine, Catholic University of Korea (CMMC-2015–0042–02, 2015–2017), which adhered to the regulations set under the Korean National Animal Protection Act. The RH strain of T. gondii has been provided by the National Veterinary Research and Quarantine Service.
Cell line and parasite
ARPE-19 cells (ATCC® CRL-2302TM, Manassas, Virginia, USA) were maintained in Dulbecco’s Modified Eagle Medium Nutrient Mixture F-12 (DMEM/F12, Invitrogen, Carlsbad, California, USA) containing 2 μM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml fungizone and 10% fetal bovine serum (FBS, Gibco Life Technologies, Grand Island, New York, USA). Tachyzoites of the RH strain of T. gondii were intraperitoneally injected into BALB/c mice, and peritoneal exudates were collected at the 4th day with Dulbecco’s PBS (DPBS) (Invitrogen).
Drugs and antibodies
Total 11 TKIs were tested in this study. DMSO was used as a solvent for the TKIs and also as the negative vehicle control. Pyrimethamine 5 μM was used for positive control. Afatinib (BIBW2992), AG1478, dacomitinib (PF299), erlotinib (OSI-420), gefitinib (ZD1839), lapatinib, neratinib, AZD9291 (osimertinib), pelitinib, and nintedanib (BIBF 1120) were purchased from Selleck Chemicals (Houston, Texas, USA). Dimethyl sulfoxide (DMSO), pyrimethamine, and sunitinib malate (SU 11248) were purchased from Sigma Aldrich (St. Louis, Missouri, USA).
Bovine serum albumin was purchased from Bovogen Biologicals (Melbourne, Australia). Antibodies against β-actin were purchased from Cell Signaling Technology (Beverly, Massachusetts, USA). FITC-conjugated anti-mouse IgG antibody, TRTIC-conjugated anti-rabbit IgG antibody, and horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibodies were purchased from Sigma Aldrich. Mouse Tg563 monoclonal antibody was cloned in our laboratory. PDCD4 (D29C6) XP® anti-rabbit monoclonal antibody was purchased from Cell Signaling.
Assay of TKIs on intracellular multiplication of T. gondii
ARPE-19 cells were plated on 12 mm cover glasses in 24-well plates (Costar, New York, USA) at a density of 0.5×105 cells/0.5 ml/well. After 24 hr, fresh tachyzoites were added to the plates at a density of 5.0×105 T. gondii/0.5 ml/well. After 1 hr of infection, non-invading parasites were washed away with pre-warmed DPBS and refilled with fresh pre-warmed medium containing 10% FBS.
Incremental doses of each TKI were applied. The drugs were added 1 hr after infection and at 24-hr intervals post-infection until 72 hr. At the indicated time point of post-infection, the cells in the wells were fixed with ice-cold methanol (Merck, Darmstadt, Germany) for 5 min and stained with Giemsa solution (Sigma Aldrich). Cells on cover glasses were observed under 400×objective fields. The number of parasites per parasitophorous vacuole (PV) was counted in each stained well at 5 random high-power fields (×400). Each experiment was performed in triplicate.
Effects of TKIs on T. gondii protein expression
ARPE-19 cells were plated in 6-well plates (Costar) at a density of 2.0×105 cells/3.0 ml/well. After 24 hr, fresh tachyzoites were added to the plates at a density of 5.0×105 T. gondii/0.5 ml/well. After 1 hr of infection, the TKIs were diluted to 5 μM/ml with DPBS and added to the wells after removing the former media. After 40 hr, the wells were washed with DPBS, and the cells were lysed with 100 μl/well of lysis buffer, i.e., 50 mM Tris-HCl, pH 6.8, 2% SDS, 1% β-mercaptoethanol (Sigma Al-drich), and 10% glycerol (Junsei, Tokyo, Japan), and 1% bromophenol blue.
The total cell lysates were analyzed by western blot. The cell lysates were dissolved using 12% SDS-PAGE and transferred to nitrocellulose membranes (Whatman GmbH, Dassel, Germany) by a mini-protean Tetra system (Bio-Rad, Hercules, California, USA). The membrane was incubated with 5% skim milk (Difco Laboratories, Detroit, Missouri, USA) in PBS with 0.5% Tween 20 (PBST) for 1 hr. Following a PBST wash, the membranes were incubated with primary antibodies (1:2,000 for T. gondii proteins, 1:10,000 for β-actin) in PBST with 5% skim milk at room temperature for 2 hr. The membranes were then incubated with secondary antibodies (anti-rabbit or anti-mouse IgG-horseradish peroxidase in PBST with 5% skim milk) for 2 hr. The signals were detected with an ECL Western blot kit (Millipore Corporation, Billerica, Massachusetts, USA). The assays were repeated 3 times, and each experiment was performed in triplicate.
Immunofluorescence assay
ARPE-19 cells were plated on 12 mm cover glasses in 24-well plates (Costar) at a density of 0.5×105 cells/0.5 ml/well. After 24 hr, fresh tachyzoites were added to the plates at a density of 5.0×105 T. gondii/0.5 ml/well. After 1 hr of infection, the drugs were diluted to 5 μM with DPBS and added to the wells after removing the former media. After 24 hr, the cells were fixed with ice-cold methanol (Merck, Darmstadt, Germany) for 5 min. The cells were blocked with 3% BSA, and then incubated for 1 hr with PDCD4 and GRA3 antibodies diluted at 1:200 and 1:1,000, respectively, with DPBS. Finally, FITC-conjugated anti-mouse IgG antibody and TRTIC-conjugated anti-rabbit IgG antibody diluted in 1:500 DPBS was applied. The assays were repeated 3 times, and each experiment was performed in triplicate.
RESULTS
TKIs inhibit intracellular multiplication of T. gondii in host ARPE-19 cells
Preliminary trial results enabled the TKIs to be divided into 3 groups. Group I consisted of nintedanib, AZD9291 (osimertinib), and sunitinib. The treating concentrations of sunitinib were low at 1, 5, and 10 μM. Nintedanib and AZD9291 were treated at higher concentrations of 5, 10, and 20 μM. Sunitinib was unable to inhibit the intracellular proliferation of T. gondii in concentrations up to 10 μM. Furthermore, it nearly destroyed the morphology of host cells after 72 hr of infection at 20 μM. Nintedanib (BIBF 1120) showed similar findings to that of sunitinib. It was unable to inhibit the intracellular proliferation in concentrations up to 10 μM, and nearly destroyed the morphology of host cells after 72 hr of infection at 20 μM. AZD9291 showed similar findings, too. It was unable to inhibit the intracellular proliferation in concentrations up to 20 μM, and nearly destroyed the host cells after 72 hr of infection at 20 μM (Fig. 1). TKIs in Group I were unable to inhibit T. gondii intracellular proliferation at 10 μM, which was close to 20 μM, in which host cell destruction was observed (Fig. 2A, B).
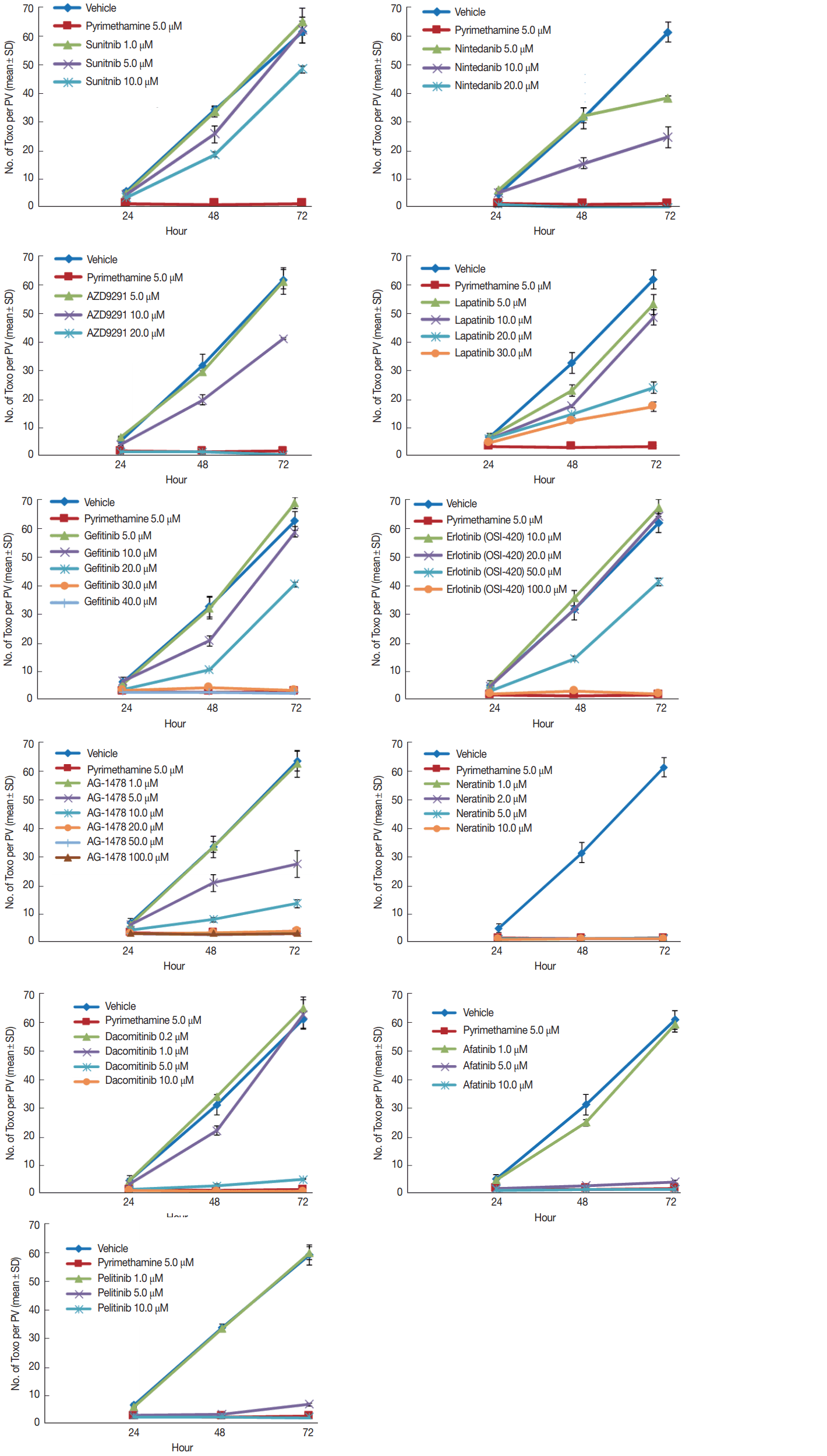
Inhibition of intracellular multiplication of T. gondii RH tachyzoites in ARPE-19 cells. The number of parasites per parasitophorous vacuole (PV) after Giemsa staining. DMSO and pyrimethamine 5 μM were used as negative and positive controls, respectively. Each experiment was performed in triplicate. Data are shown as mean±SD of the triplicate of the representative experiment.
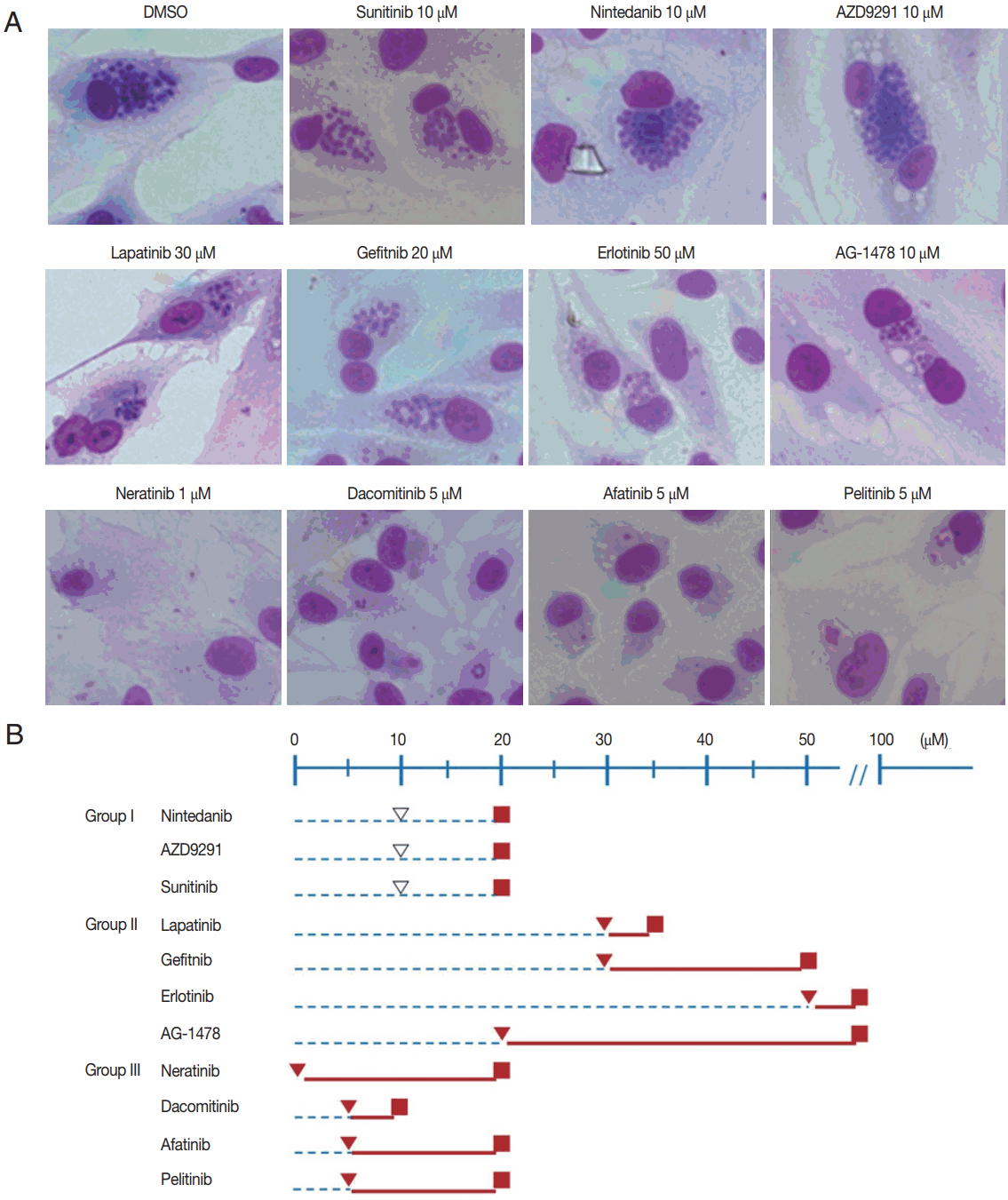
Effective and toxic concentrations of TKIs divided into 3 groups. (A) A representative result for each drug is shown. The first row shows control and group I TKIs (sunitinib, nintedanib, and AZD9291). The second and third rows show group II (lapatinib, gefitinib, erlotinib, and AG1478) and III (neratinib, dacomitinib, afatinib, and pelitinib) TKIs, respectively (Giemsa stain, ×400). (B) Group I TKIs were unable to inhibit T. gondii intracellular proliferation at 10 μM, which was close to 20 μM in which host cell destruction was observed. TKIs in Group II were able to inhibit intracellular proliferation strongly, but at relatively high concentrations of 20 μM and higher without host cell destruction at even higher concentrations. Group III TKIs were able to inhibit intracellular proliferation up to 98% equivalent to that of pyrimethamine 5 μM at very low concentrations of 1–10 μM, but host cell destruction was also observed at lower concentrations compared to Group I, at 10–20 μM. ▽ No inhibition of T. gondii proliferation, ▼ Inhibition of T. gondii proliferation up to 98% equivalent to that of pyrimethamine at 5 μM, ■ Host cell destruction. Blue dashed line represents no inhibition of T. gondii proliferation, while solid red line represents inhibition of T. gondii proliferation equivalent or higher than 98% to that of pyrimethamine at 5 μM.
Group II consists of lapatinib, gefitinib, erlotinib, and AG1478. The treating concentration of lapatinib was 5, 10, 20, and 30 μM. Gefitinib was treated similarly at 5, 10, 20, 30, and 40 μM. Erlotinib was treated at higher concentrations of 10, 20, 50, and 100 μM. AG1478 was treated at the widest range of concentrations at 1, 5, 10, 20, 50, and 100 μM. Lapatinib was unable to inhibit the intracellular proliferation of T. gondii in concentrations up to 20 μM, but at 20–30 μM it was able to inhibit up to 98% equivalent to that of pyrimethamine 5 μM. The intracellular proliferation was inhibited with gefitinib at concentrations over 30 μM, but host cells acquired significant morphological changes at these concentrations. Erlotinib was unable to inhibit the intracellular proliferation in concentrations under 100 μM, but at 100 μM, intracellular proliferation was inhibited completely, and furthermore, it induced little change in the morphology of host cells after 72 hr of infection. AG1478 at concentrations over 20 μM inhibited intracellular proliferation of T. gondii in ARPE-19 cells. At concentrations of 20 and 50 μM, it inhibited proliferation effectively while inducing little changes in the morphology of the host cells. On the other hand, at 100 μM it induced significant morphological changes (Fig. 1). In comparison to Group I, TKIs in Group II were able to inhibit T. gondii intracellular proliferation up to 98% equivalent to that of pyrimethamine 5 μM, but at relatively high concentrations of 20 μM and higher. On the other hand, host cell destruction was not observed at even higher concentrations (Fig. 2A, B).
Group III consisted of neratinib, dacomitinib, afatinib, and pelitinib. The treating concentrations of Group III TKIs were similar in being relatively low compared to the other groups. Neratinib (HKI-272) was treated at 1, 2, 5, and 10 μM. Dacomitinib was treated at 0.2, 1, 5, and 10 μM. Afatinib and pelitinib were both treated at 1, 5, and 10 μM. Neratinib (HKI-272) inhibited the intracellular proliferation of T. gondii successfully at concentrations under 1 μM. While little change in morphology was observed in host cells at 5 μM, significant changes appeared at 10 μM, and almost complete destruction occurred at 20 μM after 72 hr of infection. Dacomitinib at 5–10 μM concentration showed inhibition of intracellular proliferation. Significant changes in host cell morphology were observed at 10 μM after 72 hr of infection. Afatinib showed similar results. At 10 μM concentration it inhibited intracellular proliferation successfully, with minimal change in the morphology of host cells. But at 20 μM almost complete destruction occurred after 72 hr of infection. Pelitinib also showed similar results. Inhibition of intracellular proliferation was noted at 5–10 μM concentration. Little change in morphology was observed in host cells at 5 μM, but significant changes appeared at 10 μM, and almost complete destruction occurred at 20 μM. after 72 hr of infection (Fig. 1). Group III TKIs were able to inhibit T. gondii intracellular proliferation up to 98% equivalent to that of pyrimethamine 5 μM at very low concentrations of 5–10 μM. On the other hand, host cell destruction was also observed at lower concentrations compared to Group I, at 10–20 μM (Fig. 2A, B).
Inhibition of T. gondii protein expression is drug-dependent on the particular TKI
TgHSP90, SAG1, and GRA3 expressions were evaluated as T. gondii growth indices. Pyrimethamine 5.0 μM blocked the expression of T. gondii proteins completely as expected. Group I TKIs showed none to mild inhibition of TgHSP90 and SAG1, while GRA3 expression was moderately inhibited. Group II TKIs showed conflicting results. TgHSP90 and SAG1 expression seemed to be actually slightly enhanced, while GRA3 showed none to mild inhibition. On the other hand, AG1478 showed moderate inhibition of all 3 proteins. Group III TKIs showed almost complete blocking of protein expression comparable to pyrimethamine (Fig. 3).
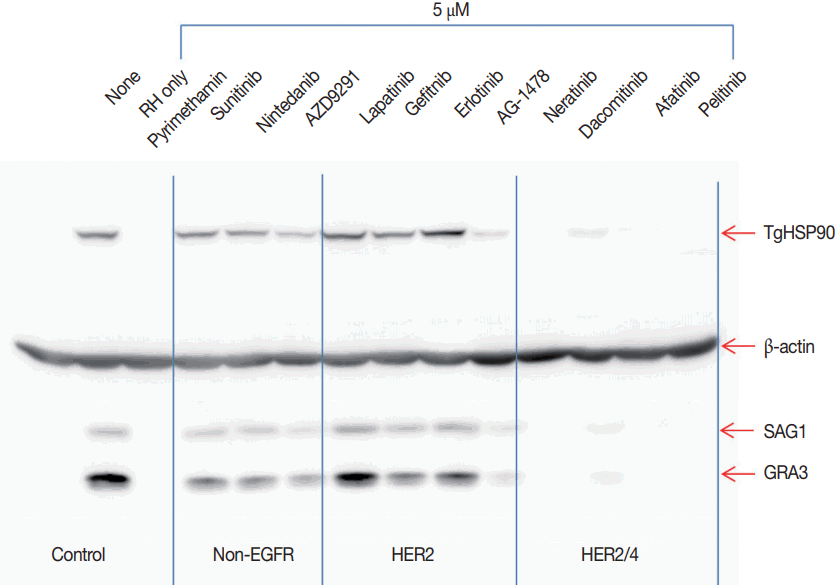
Effects of TKIs on T. gondii protein expression. Group I TKIs show none to mild inhibition of TgHSP90 and SAG1, while GRA3 expression is moderately inhibited. Group II TKIs show conflicting results. TgHSP90 and SAG1 expressions seem to be slightly enhanced while GRA3 expression shows none to mild inhibition with lapatinib, gefitinib, and erlotinib. On the other hand, another Group II TKI, AG1478, shows moderate inhibition of all 3 proteins. Group III TKIs show almost complete blocking of protein expression comparable to pyrimethamine 5.0 μM. The assays were repeated 3 times, and each experiment was performed in triplicate. A representative result is shown.
Apoptosis is drug-dependent on the particular TKI group
Group I TKIs sunitinib and AZD9291 showed similar results to that of the negative control with PDCD4 and GRA3 well localized inside the nuclei. Mild disruption of PDCD4 in the nuclei was observed with Group II TKIs gefitinib, erlotinib and AG1478. Group III TKIs neratinib, afatinib, and pelitinib showed comparable changes to that of pyrimethamine 5 uM, with complete disruption of PDCD4 and GRA3, without any localization (Fig. 4).
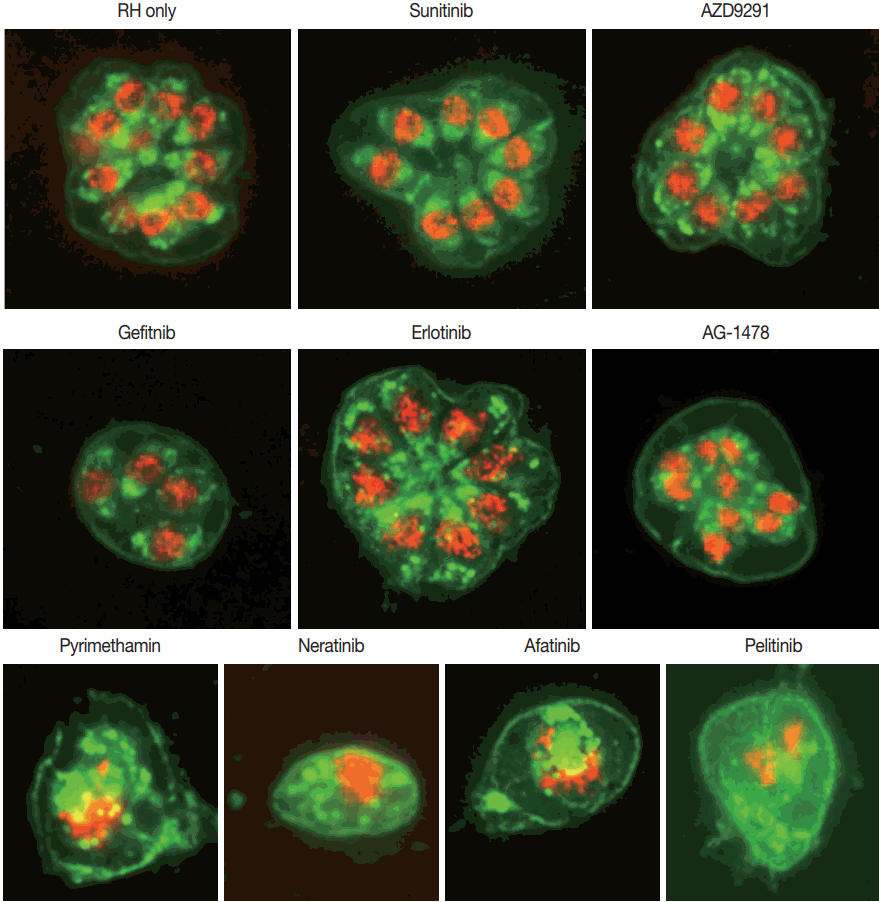
Immunofluorescence Assay. PDCD4, GRA3, and overlapping are represented in red, green, and yellow, respectively. Group I TKIs sunitinib and AZD9291 show similar results to that of the negative control with PDCD4 and GRA3 well localized inside the nuclei. Mild disruption of PDCD4 in the nuclei is observed with Group II TKIs gefitinib, erlotinib, and AG1478. Group III TKIs neratinib, afatinib, and pelitinib show comparable changes to that of pyrimethamine 5 μM, with complete disruption of PDCD4 and GRA3, without any localization. The assays were repeated 3 times, and each experiment was performed in triplicate. A representative result is shown (x1,000).
DISCUSSION
The classic regimen for treating ocular toxoplasmosis comprises of pyrimethamine and sulfadiazine with systemic corticosteroids [19]. However, this treatment modality may cause severe complications, such as Stevens-Johnson syndrome in susceptible patients and bone marrow suppression [20]. These drugs are also ineffective against bradyzoites within tissue cysts [21]. In addition, there is no effective vaccine against toxoplasmosis in humans currently.
The human epidermal growth factor receptor (EGFR/HER) family of receptor tyrosine kinases (RTKs) consists of 4 members, including EGFR (HER1, erbB-1), HER2 (erbB-2), HER3 (erbB-3), and HER4 (erbB-4) [22]. Their signaling pathways regulate cell growth, survival, adhesion, migration, and differentiation through the RAS/RAF/mitogen-activated protein kinase, phosphoinositide 3-kinase/AKT, and Janus kinase/signal transducer and activator of transcription (JAK/STAT) downstream pathways [23]. The TKIs are oral non-peptide anilinoquinazolone compounds homologous of the adenosine triphosphate (ATP), and this similarity allows them to compete for the ATP-binding domain of protein kinases preventing phosphorylation and subsequent activation of the signal transduction pathways, leading to apoptosis and decreasing cellular proliferation [24]. Additionally, TKIs target other kinase receptors due to the homology that they share with the EGFR family in the catalytic domain. The present study was conducted to evaluate the inhibitory effects of TKIs on the replication/proliferation/growth of intracellular T. gondii and their adverse effect, if any, on host ARPE19 cells.
The TKIs used in this study were mostly acquired chronologically when they became available for research. No particular selection bias was present when obtaining them. Additionally, a discussion of the individual TKIs used in this study is fundamental to our understanding of the mechanisms. Sunitinib (SU11248) is an oral inhibitor of VEGFR-1, -2, -3, PDG-FRs, KIT, FLT3, RET, and CSF-1R [25], while nintedanib (BIBF 1120) is an oral, triple angiokinase inhibitor, which acts simultaneously on 3 key receptor families involved in angiogenesis: VEGFRs, PDGFRs, and FGFRs [26]. In this regard, sunitinib and nintedanib are considered multitargeted anti-angiogenic agents [22]. AZD9291 (Osimertinib) is a third-generation EG-FR-TKI, which selectively blocks the activity of mutants but spares that of wild type EGFR [27]. Lapatinib (GW-572016) is an oral reversible TKI of EGFR and HER2 that is approved for the treatment of HER2-positive meta-static breast cancer [28].
Lapatinib inhibited cell proliferation and migration of breast cancer cell lines expressing different levels of EGFR and HER2, and its effects are mediated mainly through the HER2 pathway [29]. Gefitinib is a reversible EGFR TKI used for the treatment of non-small cell lung carcinoma with EGFR mutations. It was more potent to inhibit the proliferation of breast cancer cells with high levels of HER2 and low levels of EGFR compared to those cells with high levels of EGFR without HER2 expression [30]. Erlotinib is an orally available, reversible inhibitor of epidermal growth factor receptor (EGFR; HER1) that exhibits inhibitory activity on purified HER2 kinase at much higher concentrations [31]. AG1478 is a small molecule kinase inhibitor which acts against EGF-stimulated phosphorylation of an EGFR/HER2 chimeric receptor. It has been shown to induce the formation of inactive, unphosphorylated EGFR/HER2 heterodimers, and also inactive, unphosphorylated HER2 dimers [32] thus resulting in HER2 inhibition.
Neratinib is an oral, irreversible TKI, that is known as a pan-inhibitor because of its interaction with the catalytic domain of several EGFR family members (EGFR, HER2, and HER4), thus blocking their downstream signaling pathways [33]. Afatinib is an oral, irreversible EGFR family blocker that binds to the adenosine triphosphate (ATP)-binding pocket of EGFR, HER2 and HER4 blocking signaling pathways [34]. Pelitinib is a pan-EGFR irreversible TKI that covalently binds to EGFR, HER2, and HER4 [35]. And dacomitinib is an oral irreversible pan-EGFR TKI that acts against EGFR, HER2, and HER4 homodimers and heterodimers with a higher kinase inhibition than gefitinib/erlotinib [36].
In this study, we found that the responses of the TKIs were as varied as their target TKs. This is totally expected as the TKs themselves, even though subclassified into various families and subfamilies, groups, and such, show varied effects, both distinct and overlapping with one another. The same can be said about the toxoplasma PKs, which would be expected to have the same basic functions even if they were not eukaryotes. Adding to this plethora of behavior, various mutations can also obscure the findings, which is exactly what is happening when the TKIs are used in chemotherapy against cancers. Fortunately, we could find some kind of order among these variable results.
Interestingly, the results of our study made it possible to divide the TKIs into 3 groups. After reviewing the literature, we realized that the 3 groups were classified according to 2 criteria: action against the EGFR family and action against HER2. Group I consisted mainly of non-EGFR TKIs, group II was anti-HER2, and group III consisted of anti HER2/HER4 TKIs.
Group I consisted of nintedanib, osimertinib (AZD9291), and sunitinib. TKIs in this group were unable to inhibit T. gondii intracellular proliferation, and at a slightly higher concentration destroyed the host cells. It is probable that intracellular proliferation depends on PKs that are not influenced by non-EGFR TKs, as sunitinib and nintedanib are considered multi-targeted anti-angiogenic agents that act against VEGFRs, PDG-FRs and FGFRs.
Osimertinib (AZD9291) presented an interesting case. Even though classified as Group I, osimertinib is a third-generation EGFR-TKI. As such, it should have shown results comparable to group II or III. A reason for the difference may lie in its mechanism of action, as it blocks the activity of mutants, and is inactive against wild type EGFR. Although not the same, the corresponding ortholog T. gondii TK may be similar to wild type EGFR. This finding was similar to that of Muniz-Feliciano et al. [8].
Group II consisted of lapatinib, gefitinib, erlotinib, and AG1478. The TKIs were able to inhibit T. gondii intracellular proliferation up to 98% equivalent to that of pyrimethamine 5 μM, but only at relatively high concentrations of 20 μM and higher. On the other hand, host cell destruction was not observed at even higher concentrations. All TKIs in Group II are HER2 inhibitors.
EGFR is regulated by at least 7 distinct peptide ligands [37], but HER2 has no known direct activating ligand. It may function primarily as a coreceptor (or heterodimerization partner) for other HER receptors that do have ligands. Therefore HER2 seems to have a high level of constitutive (ligand-independent) activity [38]. On the other hand, HER2 is always poised to homodimerize or to form heterodimers with ligand-activated forms of other HERs, but HER2 homodimerization is rather weak. This suggests that HER2 heterooligomerizes with other HER more efficiently than it can homodimerize [39].
So theoretically, a TKI with prevalent anti-HER2 activity could inhibit the unknown T. gondii PK because of its HER2 characteristic. On the other hand, a higher concentration would be required. Although interruption of EGFR function with specific TKIs may disrupt EGFR-HER2 cross-talk, resulting in HER2 inactivation [40], the inhibitory effect would be limited because of other HER characteristics. A limited activity against HER2 would also cause less damage to the host cell. At first glance, HER2 inhibitors seem promising as anti-toxoplasma drugs, but the high concentrations required for inhibition would limit its usefulness.
Group III consists of neratinib, dacomitinib, afatinib, and pelitinib. TKIs in this group were able to inhibit T. gondii intra-cellular proliferation up to 98% equivalent to that of pyrimethamine 5 μM at very low concentrations of 5–10 μM. On the other hand, host cell destruction was also observed at lower concentrations compared to Group I, at 10–20 μM. TKIs in Group III are EGFR, HER2, HER4 inhibitors.
The signaling characteristics of the 4 HERs are strongly interdependent [39], and in the case of EGFR and HER4, they may readily homodimerize efficiently and form heterodimers with HER2 [41]. But as mentioned above in regard to Group I, wild type EGFR seems unaffected by these TKIs, so inhibition against HER2/HER4 seem most probable. The results using these TKIs are comparable with a former study that was performed using afatanib [4]. Because of its double action on HER2/HER4, group III TKIs would be very efficient at low concentrations, but its toxic concentration would also be low. In this respect, neratinib seems very promising as a candidate drug against toxoplasmosis, as its therapeutic window is the very large. A significant factor that may have contributed to the varied responses observed among TKIs may have resulted from the duration of the experiment, as it was performed for 72 hr. The stability of the individual TKIs also needs to be addressed. On the other hand, the results were fairly consistent within each group.
TgHSP90 contributes to bradyzoite development, and also invasion and replication of T. gondii in host cells. SAG1 is a major surface antigen and main target of the host immune response. Although accounting for only 3–5% of total T. gondii protein, most antibodies are reactive against SAG1 during infection. SAG1 promotes invasion of tachyzoites into host cells and allows tachyzoite binding to host receptors. Thus, these 2 proteins are mainly involved in the invasion and proliferation of T. gondii. In contrast, GRA3 is located inside the dense granules of T. gondii, and becomes associated with the PVM and extensions of the PVM that protrude into the cytoplasm. Modulation of intracellular calcium concentration with GRA3-CAMLG (calcium modulating ligand) interaction leads to the inhibition of host cell apoptosis for the long-term residence of invading intracellular parasites [42].
Group I TKIs sunitinib, nintedanib, and AZD9291 showed none to mild inhibition of TgHSP90 and SAG1, while GRA3 expression was moderately inhibited. These TKIs may theoretically induce host cell apoptosis and control chronic toxoplasmosis. Group II TKIs lapatinib, gefitinib and erlotinib showed conflicting results. TgHSP90 and SAG1 expression seemed to be slightly enhanced, while GRA3 showed none to mild inhibition. On the other hand, another Group II TKI, AG1478 showed moderate inhibition of all 3 proteins. This difference may be attributed to the mechanism of AG1478, as it induces the formation of inactive hetero- and homodimers, and does not inhibit HER2 directly. Another reason for this varied response may lie in the complex behavior of HER2 itself. And Group III TKIs neratinib, dacomitinib, afatinib, and pelitinib showed almost complete blocking of protein expression comparable to pyrimethamine. This was consistent with the findings related to the inhibition of intracellular proliferation. These TKIs may theoretically may inhibit both acute invasion and also induce host cell apoptosis, thereby controlling chronic toxoplasmosis. Programmed cell death 4 (PDCD4) functions as a tumor suppressor and a translation inhibitor. The loss of PDCD4 protein by apoptotic stimuli is a critical cause that induces apoptosis in several cells [43]. Group I TKIs sunitinib and AZD9291 showed similar results to that of the negative control, with PDCD4 and GRA3 well localized inside the nuclei. Mild disruption of PDCD4 in the nuclei was observed with Group II TKIs gefitinib, erlotinib and AG1478. Group III TKIs neratinib, afatinib, and pelitinib showed comparable changes to that of pyrimethamine 5 uM, with complete disruption of PDCD4 and GRA3, without any localization. Direct T. gondii destruction induced by interferon seems plausible.
Exactly how TKIs suppress T. gondii replication and proliferation remains unclear, but several mechanisms and pathways have been proposed [4,8]. EGFR activation by T. gondii, and subsequent autophagy blocking prevent killing of the parasite [8], and our study agrees in the possibility of an unknown T. gondii PK that seems to have HER2/HER4 properties. T. gondii modifies host cell processes and render the cells unresponsive to IFN-γ, thus allowing the parasite to survive in a hostile environment maintained by immunocompetent hosts. The mechanism responsible for this seems to be the inhibition of IFN-γ induced primary response genes, but the particular effector secreted from T. gondii is unknown [44]. This unknown effector may or may not be the same as the PK we have been targeting in this study. Additionally, the effector responsible seems likely to act directly on DNA-bound tyrosine-phosphorylated STAT1 [44], raising the possibility that a TKI may indeed influence immune-modulation.
Further studies are deemed necessary to determine the actual target site, pathway and mechanism for these and other TKIs that may be developed further on. It would also facilitate the development of novel drugs against toxoplasmosis.
In conclusion, HER-2/4 inhibitors seem to have a direct toxic effect against T. gondii, and may play an important role in the inhibition of its intracellular proliferation with minimal adverse effect on host cells. Indeed, with continuous development of this kind of inhibitors in cancer-related fields currently, an effective treatment modality for toxoplasmosis in the future seems most probable.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (no. 2017RIA2B4004594).
Notes
CONFLICT OF INTEREST
We have no conflict of interest related to this study.