Antimalarial Activity of C-10 Substituted Triazolyl Artemisinin
Article information
Abstract
We synthesized C-10 substituted triazolyl artemisinins by the Huisgen cycloaddition reaction between dihydroartemisinins (2) and variously substituted 1, 2, 3-triazoles (8a-8h). The antimalarial activities of 32 novel artemisinin derivatives were screened against a chloroquine-resistant parasite. Among them, triazolyl artemisinins with electron-withdrawing groups showed stronger antimalarial activities than those shown by the derivatives having electron-donating groups. In particularly, m-chlorotriazolyl artemisinin (9d-12d) showed antimalarial activity equivalent to that of artemisinin and could be a strong drug candidate.
The naturally occurring sesquiterpene endoperoxide artemisinin (1), which was isolated from Artemisia annua L., [1] serves as an interesting lead compound in the development of many drugs [2]. Artemisinin (1) is an important lead compound in the development of antimalarial drugs, especially against chloroquine-resistant strains of Plasmodium falciparum [3–6]. In addition, artemisinin (1) and its reductive product, dihydroartemisinin (2), have been used to develop novel anticancer agents [7–9]. To overcome the limitation of the poor water- and oil-soluble nature of 1, many trials have been conducted to synthesize artemisinin derivatives such as acetal-type artemisinin derivatives (3) from dihydroartemisinin (2) [10–13] and nonacetal-type derivatives (4) [14,15]. We also synthesized thiophenyl- (5) and benzenesulfonyl-dihydroartemisinin (6) and evaluated their antimalarial activities [16]. By adding a sulfidyl or sulfonyl group on the C-10 anomeric center of 1 (Fig. 1), we observed an improvement in antimalarial activity [16]. We synthesized novel artemisinin derivatives as shown in Fig. 1 because the acetal group at the C-10 position of artemisinin (1) confers chemical and biological instability [17] and causes neuronal toxicity [18]. Previous reports on its structure-activity relationship (SAR), reveal an unmet need for structural derivatization of artemisinin to facilitate novel antimalarial drug discovery. In particularly, Haynes recently reported that artemisone (10-alkylaminoartemisinin), N-sulfonyl-11-azaartemisinin, and N-carbonyl-11-azaartemisinin, having nitrogen groups at the C-10 position, showed increment of antimalarial property [19,20].
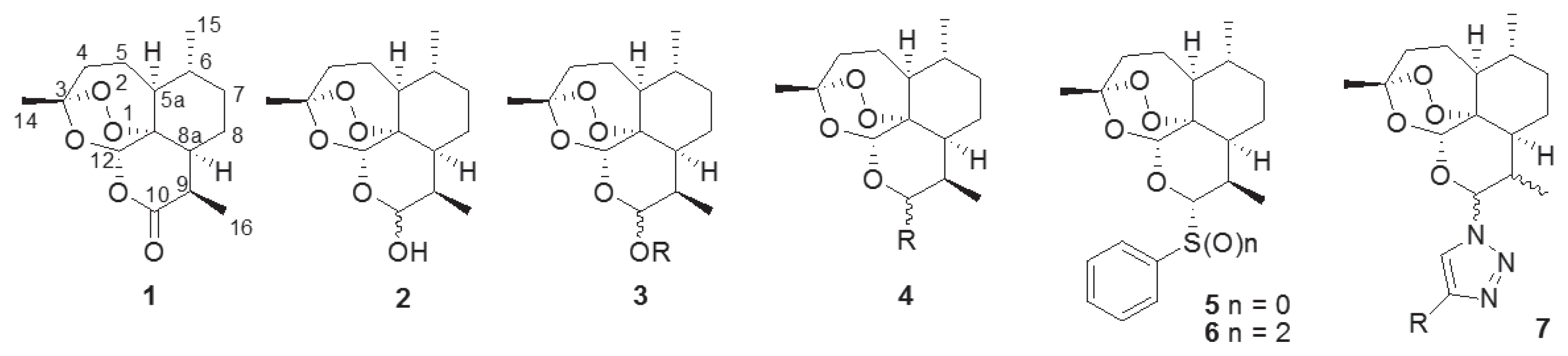
Structure of artemisinin and artemisinin derivatives with diverse functional group.
Consequently, we could safely assume that the introduction of additional nitrogen groups at the C-10 position of 1 will afford novel artemisinin derivatives with improved antimalarial activities. Therefore, we designed and synthesized various 10-substituted triazolylartemisinins (7) and tested their antimalarial activities against chloroquine-resistant P. falciparum (50005=FCR-3/Gambia subline F-86).
Novel 10-substituted triazolyl artemisinin analogs (9a–9h, 10a–10h, 11a–11h, and 12a–12h) were obtained by the Huisgen cycloaddition reaction between dihydroartemisinins (2) and variously substituted 1, 2, 3-triazoles (8a–8h) in the presence of an acid catalyst, BF3Et2O, at room temperature. By altering the reactant stoichiometric ratio, the various 10-substituted triazolyl artemisinin analogs were synthesized. A reactant stoichiometric ratio of 1:1 for dihydroartemisinin (2) and triazoles (8a–8h) yielded 3 kinds of diastereomers (9, 10, and 11). However, when the ratio was changed to 1:3, compound 12 was obtained instead of 9. The stereochemistry of compounds (12a–12h) were different from that of compounds with the C-9 methyl group (9a–9h) [21–23]. By using the above 2 different reaction conditions, 32 novel artemisinin derivatives having a substituted triazole group were obtained.
The culture-adapted chloroquine-resistant strain of P. falciparum, FCR-3/Gambia subline F-86, obtained from ATCC [24,25]. Further RPMI 1,640 medium containing hypoxanthine supplemented with HEPES buffer, sodium bicarbonate, human A serum, glutamine, gentamicin, and uninfected human oerythrocytes was used.
The in vitro antimalarial assays were conducted by modifying of the semi-automated microdilution technique described by Desjardins et al. [26] and the technique based on radiolabeled [H3] hypoxanthine incorporation described by Delhaes et al. [27]. Antimalarial activity testing of the synthetic library was carried out in 96-well microtiter plates. Stock solutions of each of the test compounds were pre-diluted with complete culture medium (RPMI 1,640 supplemented with 10% pooled human A serum), and were serially diluted 2fold in duplicate. The final concentrations ranged from 1.96 to 250 nmole/L for artemisinin and its derivatives from 11.2 to 1,435 nmole/L for chloroquine. After addition of a suspension of parasitized erythrocytes in complete culture medium (200 per well, 0.7% initial parasitemia with a majority in ring stages, 1.8–2% hematocrit) and [H3] hypoxanthine (1 μl per well; TRK74, Amersham, UK), the test plates were incubated at 37°C for 24 hr in an atmosphere of 5% O2, 5% CO2, and 90% N2. Proliferation of the parasites was evaluated after the incorporation of radiolabeled [H3] hypoxanthine into the nucleic acids of parasites and measurement in a liquid scintillation spectrometer (Packard Instrument Co., Downers Grove, Illinois, USA). Half maximal inhibitory concentration (IC50) values refer to molar concentrations of a drug causing 50% reduction in [H3] hypoxanthine incorporation, compared to that in drug-free control wells. IC50 values were estimated by linear regression analysis of log-dose-response curves.
Initially, we tested the antimalarial activities of chloroquine and artemisinin, and considered them as positive controls against the chloroquine-resistant parasite (50005=FCR-3). We then compared the potencies our synthetic artemisinin derivatives having a substituted triazole group, with those of the positive controls. Due to the chloroquine-resistant nature of the parasite, we observed a low potency for chloroquine (IC50 =101.2±25.2 nM) compared to that observed for artemisinin (IC50=1.4±0.52 nM). The potencies of antimalarial activities varied greatly depending on the structure of the triazole groups and the stereochemistry of the compounds as shown in Table 1. In relation to the variation in potency due to the structural change of the triazole groups, the most potent compounds were the artemisinin derivatives having the m-chlorophenyl tertrazole group. The IC50 values for compounds 9d, 10d, 11d, and 12d were 2.5, 4.2, 1.3, and 3.7 nM, respectively. Due to the electron-withdrawing nature of the chloride group in compounds 9d–12d, we expected compounds 9c–12c, 9e–12e, and of 9h–12h, having similar electron-withdrawing fluoride group to also show potent antimalarial activities. However, among 9c–12c, only 2 diastereomers (10c and 11c) showed antimalarial activities with similar potencies to those of the most potent antimalarial candidates (9d–12d) among the synthetic substituted triazolyl artemisinin derivatives and artemisinin. Compounds of 9e–12e, having an electron-withdrawing trifluoromethyl group (−CF3), exhibited moderately potent antimalarial activities that were nevertheless not as potent as those of compounds 9d–12d and artemisinin. However, compounds 9h–12h, having the strongest electron-withdrawing group (−F), exhibited a weak antimalarial activity. When considering the effect of a functional group attached on triazole ring, its electron (electron-donating or -withdrawing character) plays an important role in enhancing antimalarial activity. Substituted triazolyl artemisinin compounds with electron-withdrawing groups (−F, −Cl, and −CF3) showed stronger antimalarial activities than those of artemisinin derivatives with electron-donating groups (−H, −CH3, −C5H11, and −OCH3). To improve the antimalarial activities of compounds with substituted triazolyl group in our synthesis library, we suggest synthesis of substituted triazolyl artemisinins that have o-Cl and/or p-Cl, and other electron-withdrawing groups such as nitro, carbonyl, and nitrile groups. Further, while considering stereochemistry including regiochemistry, stereochemistry is not an important contributor to the functional group effects. As shown in Fig. 2, we synthesized four kinds of substituted triazolyl artemisinins by varying the stereochemistry at the C-9 and C-10 positions of artemisinin and by varying the regiochemistry of the triazole group under the 2 reaction conditions. Among the 4 kinds of stereoisomers and regioisomers synthesized, the most potent isomer was 9β, 10α compound (11). Fortunately, the yield of the reaction is high, and it is easy to obtain the amount of substance necessary for carrying out in vivo experiments.

Antimalarial activity of C-10 substituted triazolyl artemisinin against chloroquine-resistant parasite
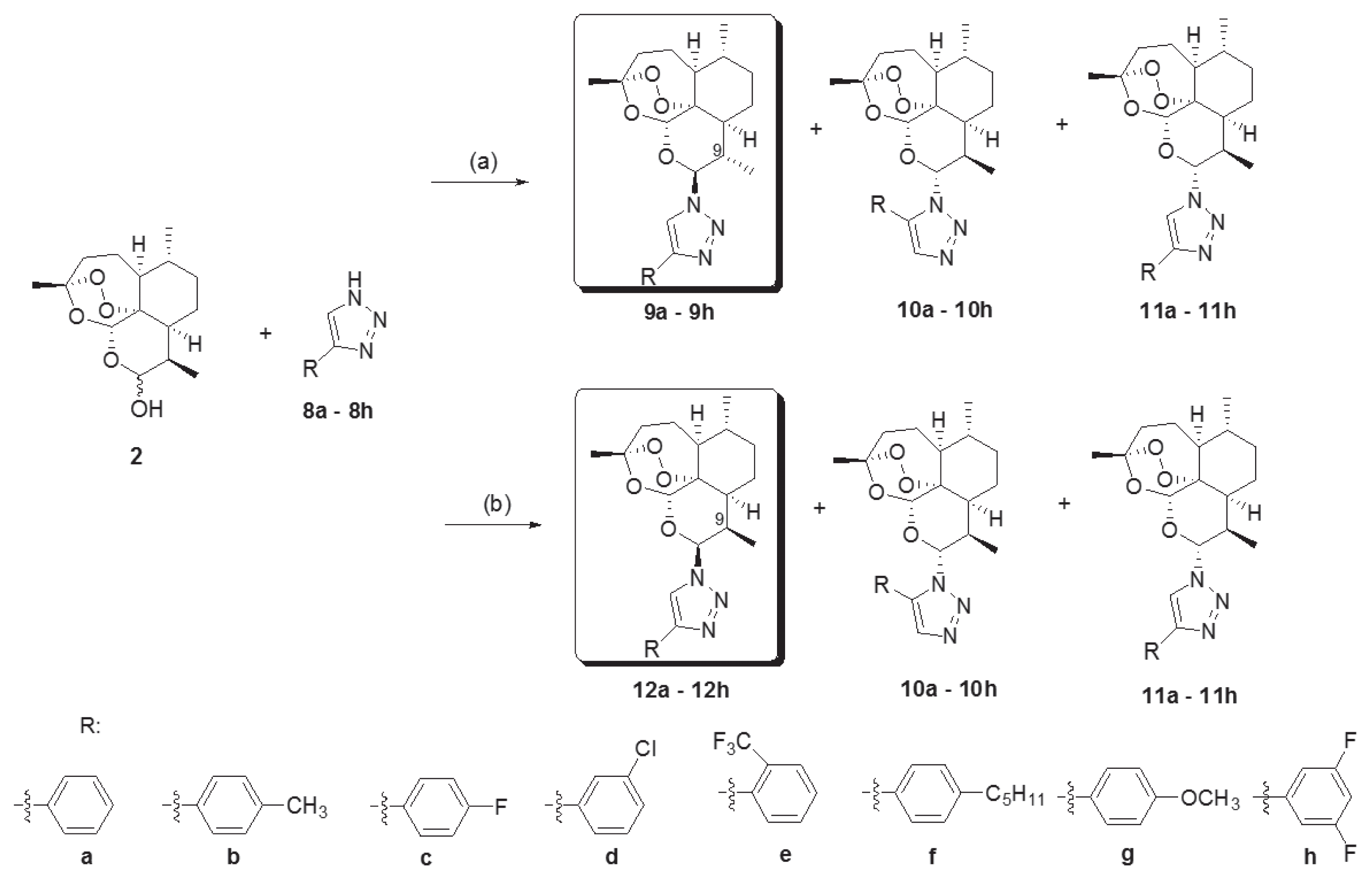
Reagents and conditions. (a) Triazole (8a–8h, 1 eq), BF3Et2O (0.8 eq) methylene chloride, rt, 24h. (b) Triazole (8a–8h, 3 eq), BF3Et2O (0.8 eq) methylene chloride, rt, 24h.
In conclusion, by the Huisgen cycloaddition reaction between dihydroartemisinins (2) and variously substituted 1, 2, 3-triazoles (8a–8h), we synthesized 4 kinds of stereoisomers and regioisomers of C-10 substituted triazolyl artemisinins under 2 reaction conditions. Among all the library compounds that we tested, m-chlorotriazolyl artemisinin (9d–12d) and o-trifluoromethyltriazolyl artemisinin (9e–12e) showed potent antimalarial activities against chloroquine-resistant parasite strain (50005=FCR-3). When considering the preliminary structure-activity relationship (SAR), electron-withdrawing property of the phenyl group attached to the triazole group plays a more important role in increasing the antimalarial activity than that played by stereochemistry. In particulary, the m-chlorotriazolyl derivatives of artemisinin (9d–12d) promises to be a strong drug candidate for novel antimalarial drug discovery.
ACKNOWLEDGMENT
This work was supported by National Research Foundation of Korea (NRF no. 2015M1A5A1037230) and by the research fund of Catholic Kwandong University.
Notes
CONFLICT OF INTEREST
We have no conflict of interest related to this work.