Genetic Diversity of Ascaris in China Assessed Using Simple Sequence Repeat Markers
Article information
Abstract
The giant roundworm Ascaris infects pigs and people worldwide and causes serious diseases. The taxonomic relationship between Ascaris suum and Ascaris lumbricoides is still unclear. The purpose of the present study was to investigate the genetic diversity and population genetic structure of 258 Ascaris specimens from humans and pigs from 6 sympatric regions in Ascaris-endemic regions of China using existing simple sequence repeat data. The microsatellite markers showed a high level of allelic richness and genetic diversity in the samples. Each of the populations demonstrated excess homozygosity (Ho<He, Fis>0). According to a genetic differentiation index (Fst=0.0593), there was a high-level of gene flow in the Ascaris populations. A hierarchical analysis on molecular variance revealed remarkably high levels of variation within the populations. Moreover, a population structure analysis indicated that Ascaris populations fell into 3 main genetic clusters, interpreted as A. suum, A. lumbricoides, and a hybrid of the species. We speculated that humans can be infected with A. lumbricoides, A. suum, and the hybrid, but pigs were mainly infected with A. suum. This study provided new information on the genetic diversity and population structure of Ascaris from human and pigs in China, which can be used for designing Ascaris control strategies. It can also be beneficial to understand the introgression of host affiliation.
INTRODUCTION
Ascariasis in humans is caused by infection with the soil-transmitted giant roundworm Ascaris lumbricoides. Approximately 760 million people are infected with this roundworm worldwide [1]. Although the majority of infections occur in developing countries, especially in Asia and Africa, cases have been reported in developed countries such as Japan, the United States, and Denmark [2–4]. The closely related parasite Ascaris suum mainly infects pigs [5]. A. lumbricoides and A. suum have similar transmission cycles and morphologies [6]. Cross-transmission of certain haplotypes and hybrids have been observed [7].
There has been considerable controversy about the taxonomic relationship between A. lumbricoides and A. suum. One view is that A. lumbricoides and A. suum are both valid species that persist in separate transmission cycles with limited gene flow [8]. The second is that A. lumbricoides and A. suum are different species existing in host-specialist parasite populations, but that there are some cross-infection and hybrids [7]. The third is that A. lumbricoides and A. suum are actually the same species [6, 9,10].
Many molecular techniques, such as isoenzyme restriction fragment length polymorphism RFLP analyses of nuclear genes and mitochondrial DNA sequences have been used to study the genetic diversity and population structure of Ascaris [9,11]. However, there are limitations in the analyses of genetic diversity and population structure using markers. For example, nuclear markers and isoenzymes have a low frequency of polymorphisms and mitochondrial DNA is maternally inherited and reflects the evolution of females rather than of the entire population [12]. In addition, the use of a single molecular marker can provide results that are misleading [13]. Therefore, new molecular techniques should be applied to investigate the genetic diversity and population structure of Ascaris. Microsatellite markers are regarded as an ideal tool for the examination of populations because they are co-dominantly inherited, easily amplified, and abundant [14]. Anderson and colleagues first applied microsatellite markers to understand mating of A. lumbricoides [15]. Then, Criscione and colleagues [16] developed and assessed 35 microsatellite markers for Ascaris, providing candidate markers for investigation of molecular epidemiology [17,18], mating patterns [5], cross-infection and hybridization [7,16], and genetic diversity [19,20].
Ascariasis is not considered a high-priority disease globally. However, ascariasis is still a public health problem in China [7] and additional prevention and control strategies are needed. To inform these strategies, the fine-scale genetic structure and microepidemiology of Ascaris in China must be surveyed. The genetic diversity and population structure of Ascaris in China has been examined using nuclear and mitochondrial markers, but rarely using microsatellite markers [8,11,21].
Previously, we determined the frequency and distribution of cross-infection and hybridization of human and pig Ascaris in sympatric populations in China [7]. In this paper, we re-analyzed these data (1) to determine the genetic diversity and structure of 12 Ascaris populations in China using multiple polymorphic microsatellite markers, (2) to apply traditional epidemiological models to population genetics, and (3) to better understand the relationship between A. lumbricoides and A. suum.
MATERIALS AND METHODS
Specimens and data collection
Specimens were collected from 12 Ascaris populations in humans (H) and pigs (P) in 6 provinces of China (Yunnan [YN], Hainan [HN], Jiangxi [JX], Xinjiang [XJ], Liaoning [LN], and Qinghai [QH]) described with 3-letter codes to indicate these sources. Twenty microsatellite loci in 258 samples were amplified according to methods described by Zhou et al. [7].
Microsatellite and genetic diversity analyses
The frequencies of null alleles were calculated using Micro-Checker ver. 2.2.3 [22]. The number of alleles (Na), allelic richness (Ar), and inbreeding coefficient (Fis) per population were computed using Fstat 2.9.3.2 [23]. GENETIX ver. 4.05.2 was used to calculate expected heterozygosity (He) and observed heterozygosity (Ho) per population [24]. Genepop ver. 4.0.7 was used to test linkage disequilibrium and Hardy-Weinberg equilibrium (HWE) with Bonferroni correction [25].
Population genetic structure analysis
Pairwise fixation index values (Fst) with ENA (excluding null alleles) correction was inferred using the FreeNA package [26]. Population genetic structure was analyzed in Structure ver. 2.3.4 using a Bayesian algorithm [27]. We used admixture ancestry and correlated allele frequency models with 20 runs and 100,000 Markov Chain Monte Carlo (MCMC) repetitions after a burn-in period of 100,000 interactions for each group number K. The appropriate K-value was determined using the method proposed by Evanno et al. [28] in the Structure Harvester program. Arlequin ver. 3.11 was used for a hierarchical analysis of molecular variance (AMOVA) [29].
RESULTS
Microsatellite variation and genetic diversity
The Micro-Checker analysis indicated that all alleles were null except at loci ALGA44, ALAC32, L017-est, and ALTN01. The frequencies of null alleles per locus ranged from 0.0000 to 0.2087 (Supplementary Table S1).
There were 2,435 alleles at 20 microsatellite loci in the 12 populations. The number of alleles observed (Na) per population varied from 143 to 276 (Table 1) and allelic richness (Ar) ranged from 6.7037 to 10.2161. Among sympatric populations of Ascaris from human and pigs, the number of alleles and allelic richness were greater in the populations from humans. Expected heterozygosity (He) and observed heterozygosity (Ho) ranged from 0.6209 to 0.7867 and 0.5299 to 0.6575, respectively. In all populations, the expected heterozygosity was greater than the observed heterozygosity and the Fis per population varied from 0.153 to 0.298.

Genetic diversity in 12 populations of Ascaris
The genotypic disequilibrium for each pair of 20 microsatellite loci was examined and significant linkage disequilibrium was identified in 6 loci: loci ALGA20 and ALGA15, ALGA32 and ALAC32, ALAC32 and L010, ALGA32 and ALAC01, ALGA15 and ALAC01, and L010 and ALAC01 Supplementary Table S2).
Microsatellite analysis revealed that there were 148 population-loci that deviated significantly from HWE at 20 microsatellite loci in the 12 Ascaris populations (Supplementary Table S3). When all loci were considered, deviation from HWE was detected in all populations (Supplementary Table S4).
Population differentiation
The pairwise Fst in the 12 populations ranged from 0.0065 to 0.1620, with a global value of 0.0567. The greatest genetic variation was observed in the populations HN-H and JX-P, whereas the least differentiation was in XJ-H and JX-H. The Fst in human Ascaris populations ranged from 0.0065 to 0.0496, with a mean of 0.0263. The Fst in Ascaris populations in pigs ranged from 0.0478 to 0.1271, with an average of 0.0810. The Fst in Ascaris populations in humans and pigs ranged from 0.0242 to 0.1620, with a mean of 0.0819 (Table 2).

Pairwise Fst values (below the diagonal) and probabilities (above the diagonal) over 20 loci in the 12 Ascaris populations
Genetic relationships and population genetic structures
The 258 Ascaris specimens were further examined for genetic relationships using a Bayesian model in the software Structure. Possible population K-values from 1 to 12 were analyzed. The Bayesian clustering analysis showed that as K increased, InP (D) increased, then decreased (Supplementary Fig. S1). Subsequently, we calculated the relationship between Δ K and K, using a method described by Evanno et al. [28]. The clear maximum value of Δ K was at K=3, which indicated that the 12 populations could be differentiated into 3 groups (Supplementary Fig. S2). These groups are shown with red, green, and blue in Figure 1. Populations JX-P, XJ-P, QH-P, and LN-P had the highest membership coefficients (JX-P, 0.915; XJ-P, 0.928; QHP, 0.872; LN-P, 0.800) in the second cluster (green). The majority of individuals from populations XJ-H, YN-H, and YN-P belonged to cluster 1 (red), whereas most individuals from JXH, HN-H, and LN-H belonged to cluster 3 (blue) (Supplementary Table S5).
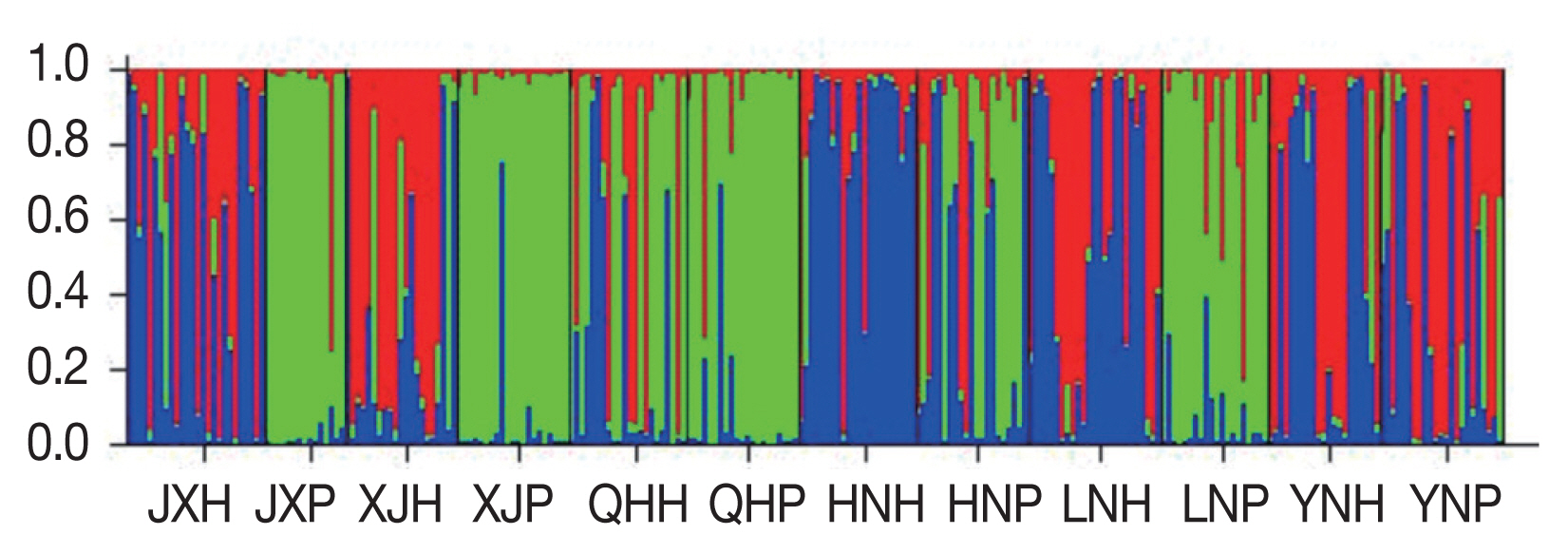
Population genetic structure at K=3 based on microsatellite data from Ascaris sampled in different geographical regions. H, human Ascaris; P, pig Ascaris; JX, Jiangxi; XJ, Xinjiang; QH, Qinghai; HN, Hainan; LN, Liaoning; YN, Yunnan.
The AMOVA indicated greater genetic differentiation within populations than between populations. The genetic difference between Ascaris from pigs and Ascaris from humans was 1.9854%, with 3.9398% between populations in a group and 94.0749% within populations. The structure analysis of the 3 genetic groups revealed limited variation between groups (1.9557%) and considerable variation within populations (94.3743%) (Table 3).

Analysis of genetic variance in the Ascaris populations
DISCUSSION
Microsatellite DNA molecular markers are widely used in population genetics research because of their high frequency of polymorphisms [30]. However, ubiquitous null alleles can cause allele numbers and frequencies to be underestimated, affecting assessments of genetic diversity [31]. A null allele is an allele of one locus that is not amplified, which can affect the accuracy of certain parameters based on the proportion of heterozygotes and, in particular, the accuracy of the inbreeding coefficient. In addition, higher null allele frequencies can reduce estimates of the genetic diversity within populations, causing fixation indices to be overestimated and the extent of gene flow to be underestimated in the analysis [32]. In the present study, the frequencies of null alleles per locus ranged from 0.0000 to 0.2087 (Supplementary Table S1). Null alleles in Ascaris populations in China could be a result of inbreeding (see discussion below). Inbreeding can accelerate the homogenization of populations, increasing the frequency of null alleles. When the null allele frequency is less than 0.2, they do not affect the accuracy of the data analysis [32]. Although there were various frequencies of null alleles in this study, accuracy of the results was not affected.
High genetic diversity was observed in the Ascaris populations at 20 simple sequence repeat (SSR) regions (Table 1). This result was similar to that based on mitochondrial data [21]. Among sympatric populations of Ascaris from human and pigs, the number of alleles and allelic richness was greater in the populations from human, which may because of cross-transmission (human infections with Ascaris from pigs) and hybridization [2–4,7,18]. In all populations, the expected heterozygosity was higher than the observed heterozygosity and Fis values were positive. The same results were obtained for populations of Ascaris in southwestern Uganda [20]. Positive Fis values indicate an excess of homozygotes [16]. The heterozygote deficiency (Ho<He, Fis>0) demonstrated in these Ascaris populations may due to inbreeding. Polyandry [5], hybridization [7], Wahlund effects [16], or null alleles [20] can also result in such a deficiency.
When genetic variation is studied at 2 or more loci simultaneously, allele frequency is insufficient to indicate the extent of genetic variation in natural populations, so the non-random association of alleles at different sites must also be considered [33]. Linkage disequilibrium refers to the non-random association of alleles at genetic loci [34]. The decay of linkage disequilibrium is affected by many factors, including genetic drift, natural selection, mutation, and gene flow [35]. Linkage disequilibrium was apparent in this study, but not all associations were identified. Significant associations between pairs of loci were also not tested in populations of A. lumbricoides from Nepal [16] and similar results were found in insect species [36].
The microsatellite analysis revealed a departure from HWE for a number of loci in the Ascaris populations (Supplementary Table S3). Taking all loci into account, all populations deviated from HWE (Supplementary Table S4). Similar results were reported for Ascaris populations in southwestern Uganda [20]. A lack of heterozygotes may be a universal phenomenon in Ascaris populations. Criscione et al. [16] also found a departure from HWE caused by heterozygote deficiency by developing and assessing microsatellite markers in A. lumbricoides. In almost all cases, a departure from HWE was also observed in southwestern Uganda [20]. Using Micro-Checker software, we detected null alleles. Thus, departures of populations from HWE might have resulted from heterozygote deficiency and/or null alleles.
Pairwise Fst values were divided into 4 categories: Fst<0.05 indicated that genetic differentiation among populations was very low; 0.05<Fst<0.15 signified moderate genetic differentiation among populations; 0.15<Fst<0.25 indicated that genetic differentiation was high; and Fst>0.25 expressed a very high level of genetic differentiation [37]. When population structuring in Ascaris from humans was investigated, there was little evidence for genetic differentiation between worm populations (Fst=0.0264) (Table 2). However, when populations from humans were compared with populations from pigs, genetic differentiation was moderate (Fst=0.0819), as it was between populations from pigs (Fst=0.0810). Differentiation between populations of Ascaris from China, measured by Fst and based on mitochondrial DNA, was previously shown to vary greatly [8]. Fst based on microsatellite data suggested that there was gene flow in Ascaris populations (either within human- or pig-derived populations or between the 2 species), similar to that shown in our previous research [21]. Despite the wide geographical area over which specimens were collected, genetic differentiation was limited and, similar to the results of previous studies [9,16], no clear host or geographical patterns were identified. There was evidence for panmixia or high gene flow between worm populations in 2 villages in southwestern Uganda based on the haplotype data and microsatellite analysis, despite their physical separation [20].
In population genetics studies, the question of how to objectively identify and divide homogeneous populations has plagued researchers [27]. This problem was solved by Structure. Then, the problem was how to objectively determine the cluster values (K) in cluster analyses. Evanno developed a ΔK method to calculate the possible values for the cluster K. The Bayesian clustering analysis showed that, with an increase in K, the values of InP (D) first increased and then decreased (Supplementary Fig. S1). This result indicated there was weak differentiation among the 12 worm populations. The optimal cluster was difficult to be determined using Pritchard’s methods based on InP (D) [27]. This may have been because, in highly mixed worm populations, a gene frequency gradient was formed. In this case, the value of K was also vague and inaccurate according to lnP (D). To determine the appropriate K value, the method of Evanno was used. In this study, the ΔK was largest at K=3 (Fig. 1). Therefore, the 12 Ascaris populations could be divided into 3 larger groups.
The result of the Structure analysis revealed that there were 3 clusters of Ascaris in China and that each cluster consisted of several populations. Populations from pigs (JX-P, XJ-P, QH-P, LN-P) constituted one group. The rest of the populations were divided into 2 genetic groups that did not represent their host or geographic distributions. The genetic structure could be explained by the hypothesis that the 3 groups represent A. suum, A. lumbricoides, and Ascaris hybrids, respectively. Animal experiments have shown that mice can be infected with eggs of A. suum; however, pigs were not susceptible to infection with eggs of A. lumbricoides [38]. In addition, humans can be infected by A. suum [2–4,18]. This information may explain why cross-infection and hybridization can occur in humans. We propose that people in China become infected with A. lumbricoides, A. suum, and a hybrid of the 2 species, but that pigs are not susceptible to this hybrid. Of course, this hypothesis requires testing with animal experiments.
The AMOVA analyses show variation in Ascaris population, similar to what was shown by Cavallero et al. [9]. Our results confirmed that gene flow between populations of Ascaris was strong and that population differentiation was weak. The genetic variation among the 3 groups was relatively small, but statistically significant (Table 3).
Based on results of Fst statistics, a Bayesian clustering analysis, and AMOVA using microsatellite data, worm population differentiation was low and gene flow was high between the populations. These findings are consistent with those of a previous Bayesian clustering study [7]. The absence of differences could be a result of complex transmission mechanisms. Although there are no confirmed drug resistance genes in Ascaris, more attention should be paid to the high degree of gene flow in Ascaris [39].
Conclusively, the results of this study provided additional insights into the genetic diversity and population structure of Ascaris from humans and pigs in China. This knowledge can be useful for treatment and control of ascariasis. It might also be beneficial in understanding the co-evolution of hosts and parasites and the introgression of host affiliation and/or drug-resistance genes between different parasite populations.
Supplementary materials
Null allele frequency per locus
Analysis of disequilibrium of pairwise loci across all populations
Hardy-Weinberg exact test at 20 simple sequence repeat loci in 12 populations of Ascaris
Table Hardy-Weinberg exact tests for each population
Proportion of membership coefficients with K=3 in each population
Relationship between lnP (D) and K-values from the STRUCTURE analysis.
Number of inferred clusters (K) with the highest probability determined by applying Evanno’s Δ K method.
ACKNOWLEDGMENTS
This work was funded by the National Natural Science Foundation of China (81460318). Thanks to Prof. Jun Rong at Nanchang University, China, for his help in data analysis. Thanks also to Prof. Robin B. Gasser at the University of Melbourne, Australia, for kindly revising the manuscript.
Notes
CONFLICT OF INTEREST
All authors declare no conflict of interest.