Abstract
Acanthamoeba spp. are free-living protozoa that are opportunistic pathogens for humans. Cysteine proteases of Acanthamoeba have been partially characterized, but their biochemical and functional properties are not clearly understood yet. In this study, we isolated a gene encoding cysteine protease of A. castellanii (AcCP) and its biochemical and functional properties were analyzed. Sequence analysis of AcCP suggests that this enzyme is a typical cathepsin L family cysteine protease, which shares similar structural characteristics with other cathepsin L-like enzymes. The recombinant AcCP showed enzymatic activity in acidic conditions with an optimum at pH 4.0. The recombinant enzyme effectively hydrolyzed human proteins including hemoglobin, albumin, immunoglobuins A and G, and fibronectin at acidic pH. AcCP mainly localized in lysosomal compartment and its expression was observed in both trophozoites and cysts. AcCP was also identified in cultured medium of A. castellanii. Considering to lysosomal localization, secretion or release by trophozoites and continuous expression in trophozoites and cysts, the enzyme could be a multifunctional enzyme that plays important biological functions for nutrition, development and pathogenicity of A. castellanii. These results also imply that AcCP can be a promising target for development of chemotherapeutic drug for Acanthamoeba infections.
-
Key words: Acanthamoeba castellanii, cysteine protease, lysosome, trophozoite, cyst
INTRODUCTION
Acanthamoeba spp. are free-living protozoa that are found not only in diverse natural environments such as soil, water and air, but also in artificial facilities including tap water systems, cooling towers, sewage and air conditioning systems [
1].
Acanthamoeba spp. are opportunistic pathogens that can cause granulomatous amoebic encephalitis, dermatitis, and amoebic keratitis in humans [
2].
Acanthamoeba has a simple life cycle consisting of trophozoites and cysts. Trophozoites are metabolically active forms, so that they actively multiply, move, and feed through active phagocytosis [
3,
4]. The resulting phagosomes fuse with lysosomes that contain acidic hydrolytic enzymes, and eventually create phagolysosomes, where the engulfed molecules are degraded. Under harsh environmental conditions, the vegetative trophozoites of
Acanthamoeba are converted to dormant cysts, which prolong their survivals for a considerable period, by a process termed encystation [
5–
7].
Cysteine proteases of parasitic organisms are known to play pivotal roles in diverse biological events such as nutrition, host cell or tissue invasion and host-parasite interactions [
8–
11]. In
Acanthamoeba, activities of cysteine proteases with different molecular sizes have been detected in both cell lysates and cultured supernatants of trophozoites, and these enzymes are thought to be involved in the proteolytic degradation of cellular components, including iron-binding proteins [
12–
18]. Until now, 2 cathepsin L-like cysteine proteases, called as AcCP2 [
19] and AhCP [
20], were identified in
Acanthamoeba spp., specifically
Acanthamoeba culbertsoni and
Acanthamoeba healyi, respectively. Cysteine proteases of
Acanthamoeba are also likely to play pivotal roles for encystation of the amoeba. E-64d, a membrane-permeable cysteine protease inhibitor, effectively inhibited encystation of
Acanthamoeba, which suggesting cysteine proteases might be essential for early phase of encystations [
16]. A cysteine protease having cathepsin B-like activity, called cyst-specific cysteine protease (CSCP), was highly expressed in encystation phase of
A. castellanii [
17]. CSCP-knockdown
A. castellanii resulted in decrease of cyst formation with a large amount of mitochondria that was not digested within autophagolysosomes. These results collectively suggested possible roles for cysteine proteases in
Acanthamoeba, but the biochemical properties and biological functions of
Acanthamoeba cysteine proteases are not clearly understood yet.
In the present study, we identified and characterized a cathepsin L-like cysteine protease of A. castellanii (AcCP). Structural and biochemical analysis of AcCP suggested that this enzyme is a typical cysteine protease, which is expressed in both trophozoites and cysts. The enzyme is likely to be multifunctional enzyme that plays important biological functions for nutrition, development and pathogenicity of A. castellanii.
MATERIALS AND METHODS
Cultivation of Acanthamoeba and induction of encystation
Acanthamoeba castellanii Castellani (ATCC #30011; Manassas, Virgina, USA) was cultured axenically in peptone-yeast-glucose (PYG) medium at 25°C. Encystation was induced as previously described [
21]. Briefly, approximately 5×10
5 cells in post-logarithmic growth phase were collected, washed with phosphate-buffered saline (PBS), and incubated in 10 ml of encystation medium (95 mM NaCl, 5 mM KCl, 8 mM MgSO
4, 0.4 mM CaCl
2, 1 mM NaHCO
3, and 20 mM Tris-HCl, pH 9.0) for 72 hr. The morphological change from trophozoites into cysts was observed by microscopy. Encystation efficiency was calculated by counting cysts after treating cells with 0.05% sarkosyl and 0.22% trypan blue, which selectively stains nonviable cells [
21,
22].
Cloning of the AcCP gene
A. castellanii mRNA was isolated from trophozoites using an Oligotex mRNA purification kit (Qiagen, Valencia, California, USA). Single-stranded cDNA was synthesized using the BD SMART™ RACE cDNA amplification kit (BD Biosciences, Palo Alto, California, USA) followed by the manufacturer’s instructions. A partial sequence of AcCP gene was amplified by PCR from cDNA using degenerate oligonucleotide primers designed based on conserved amino acids flanking the active sites of the presently known cathepsin L-family cysteine proteases. The forward primer was 5′-TGYGGWTCGTGYTGGTCATT-3′ and the reverse primer was 5′-CCAAGTGTTCYTGACYRTCCA-3′. The amplification reaction was done with a thermal cycling profile of 94°C for 4 min, 35 cycles at 94°C for 1 min, 50°C for 1 min and 72°C for 1 min, followed by a 72°C extension for 10 min. The PCR product was analyzed on 1.2% agarose gel, gel-purified and ligated into the pGEM-T Easy vector (Promega, Madison, Wisconsin, USA). The ligation was transformed into
Escherichia coli DH5α competent cells and positive clones were screened by colony PCR for the presence of plasmid with the appropriate insert. A total of 20 clones were randomly selected and the nucleotide sequences of the inserts were determined. As the result, partial gene sequence encoding for AcCP, which did not match any presently known genes encoding
Acanthamoeba cysteine proteases, was obtained. To obtain a full-length sequence of AcCP, rapid amplification of cDNA ends (RACE) procedures were performed using the BD SMART™ RACE cDNA amplification kit (BD Biosciences). Following synthesis of the first strand cDNA, the second strand cDNA was synthesized and ligated to the cDNA adaptor. The 5′- and 3′-RACE were carried out with gene-specific primers (GSPs) and an adaptor primer (AP1). GSPs were designed based on the AcCP partial sequence obtained from the PCR with degenerate primers. GSP1 for the 5′-RACE was 5′-CTCAGAGAGCGAGACCAGAGTGCCACG-3′ and GSP2 for 3′-RACE was 5′-ACTGAGAACGGCCAGGATTACTGGCTC-3′. The PCR products were gel purified, ligated, transformed and sequenced. Primary structure analysis of the deduced amino acid sequences and phylogenetic analysis were conducted with DNASTAR (DNASTAR, Madison, Wisconsin, USA), PSORT (
http://www.psort.org/) and Signal P (
http://www.cbs.dtu.dk/services/SignalP/). The phylogenetic tree was constructed using the neighbour-joining method with MEGA4 (
http://www.megasoftware.net). Bootstrap proportions were used to assess the robustness of the tree with 1,000 bootstrap replications. The gene encoding AcCP was deposited in GenBank with an accession number of EF053509.
Production of recombinant AcCP (rAcCP) protein
To produce rAcCP protein, a fragment harboring a portion of the prodomain and the entire mature domain of AcCP was amplified using the following primers: 5′-GGATCCGAGGAGTTTGTCTTCAGGTGG-3′, which harbors a 5′
BamHI site and 5′-AAGCTTTTAAGCGGTGGGGTAGGAGGC-3′, which contains a 5′
HindIII site. The purified PCR product was ligated into the pGEM-T Easy vector (Promega) and was transformed into
E. coli DH5α. The resulting plasmid DNA was digested with the appropriate restriction enzymes, ligated into the pQE-30 expression vector (Qiagen), which was then transformed into
E. coli M15 (pREP4) cells (Qiagen). The selected clones were cultured in Luria-Bertani broth supplemented with 100 μg/ml ampicillin and 50 μg/ml kanamycin at 37°C and rAcCP expression was induced with isopropyl-β-D-thiogalactoside (IPTG) at a final concentration of 0.1 mM for 3 hr at 37°C. The cells were harvested by centrifugation, suspended in 8 M urea lysis buffer and incubated overnight at room temperature. The supernatant was collected by centrifugation and the rAcCP was purified from the supernatant with nickel-nitrilotriacetic acid (Ni-NTA) agarose (Qiagen) according to the manufacturer’s instructions. Purification and purity of the recombinant protein was analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Refolding of the purified recombinant protein was performed as describe previously [
23,
24]. Ni-NTA affinity purified rAcCP (1 mg) was slowly added to 100 ml of 100 mM Tris-HCl (pH 8.0) containing 1 mM EDTA, 250 mM L-arginine, 5 mM reduced glutathione (GST), 1 mM oxidised glutathione (GSSG) and gently stirred at 4°C overnight. The sample was dialysed against 10 mM Tris-HCl (pH 7.5) and concentrated with Centricon Plus (cut-off 10 kDa; Millipore, Billerica, MA, USA). The resulting rAcCP was further processed in different pH buffers (pH 4.0 to 7.0) to analyze the optimal pH condition required for processing to fully matured enzyme as describe previously [
24]. In brief, the refolded rAcCP was incubated in different pH buffers (pH 4.0 to 7.0). The aliquots were taken at the indicated times and were analyzed by SDS-PAGE and enzyme activity assay. The enzyme activity of rAcCP was assayed fluorometrically as the hydrolysis of benzyloxycarbonyl-L-leucyl-L-arginine-MCA (ZLR-MCA; Peptide Institute, Osaka, Japan). Fluorescence was measured at excitation and emission wavelengths of 355 and 460 nm, respectively, using a Fluoroskan Ascent FL (Thermo, Vantaa, Finland).
Mouse polyclonal antibody specific for AcCP (anti-AcCP) was produced by immunizing mice with purified rAcCP protein. Briefly, purified rAcCP protein (100 μg) was intraperitoneally injected into 2 8-week-old BALB/c mice (Bionics, Seoul, Korea) with an equal volume of Freund’s complete adjuvant (Sigma, St. Louis, Missouri, USA). Before immunization, blood samples were collected from the mice for control experiments. Two boosters with rAcCP (100 μg) mixed with an equal volume of Freund’s incomplete adjuvant (Sigma) were administered at 14 and 28 days after the first injection, respectively. Blood was obtained from the heart of mice at 7 days after administration of the final booster, and anti-AcCP were obtained from the blood via centrifugation for 15 min at 4°C and analyzed its reactivity against rAcCP by Western blotting.
Biochemical properties of rAcCP protein
The optimal pH for rAcCP activity was determined in sodium acetate (pH 3.5–5.5), sodium phosphate (pH 6.0–6.5), and Tris-HCl (pH 7.0–7.5). For these experiments, 50 nM rAcCP was added to each buffer containing 5 μM Z-LR-MCA and 10 mM DTT. Appropriate pH blanks were included as controls. To examine the pH stability of rAcCP, the enzyme was incubated at 37°C in the appropriate buffer (pH 4.0–8.0), and residual enzyme activity was measured as described above. The kinetic parameters of rAcCP for Z-LR-MCA, Z-L-phenylalaninyl-L-arginine-MCA (Z-FR-MCA) and Z-L-arginyl-L-arginine-MCA (Z-RR-MCA) were determined at room temperature, using a constant amount of enzyme (30 nM) and varying concentrations of substrate in 100 mM sodium acetate (pH 4.0) containing 10 mM DTT. The concentration of rAcCP was determined by active site titration with trans-epoxy-succinyl-L-leucylamido( 4-guanidino)-butane (E-64). The kinetic constants Km and Vmax were determined using GraphPad software (GraphPad Software Inc., La Jolla, California, USA).
Degradation of human proteins by rAcCP
To assess the degradation of human proteins by rAcCP, various proteins (2 mg/ml) were separately incubated with rAcCP (100 nM) in 50 mM sodium acetate (pH 4.0) or Tris-HCl (pH 7.0) in the presence of 1 mM GSH for 3 hr at 37°C. The proteins used for hydrolysis assay were hemoglobin (from human blood), albumin (from human serum), immunoglobulin G (IgG; from human serum), IgA (from human serum) and fibronectin (from human plasma), all purchased from Sigma. After the reactions were terminated by adding reducing sample buffer, protein degradation was analyzed by SDS-PAGE.
Localization of AcCP
To determine the cellular localization of AcCP, the AcCP gene was cloned into the pGAPDH-EGFP vector, which contains an
Acanthamoeba glyceraldehyde 3-phosphate dehydrogenase (GAPDH) promoter for constitutive expression and an enhanced green fluorescent protein (EGFP) reporter gene [
25]. The AcCP gene was amplified by PCR with primers that incorporated an
NdeI restriction site at the 5′-end (5′-GGCATATGTACAAGCATGACCCGCTTAC-3′) and an
SpeI restriction site at the 3′-end (5′-GGACTAGTAGCGGTGGGGTAGGAGGC-3′), and the PCR product was cloned into the pGAPDH-EGFP. The resulted plasmids (pGAPDH-AcCP-EGFP) was then transfected to
A. castellanii trophozoites followed by the method described previously [
26]. Briefly, approximately 4×10
5 cells/well were seeded into a 6-well culture plate containing 3 ml of PYG medium and incubated at 25°C overnight. Plasmid DNA (4 μg) in 100 μl of amoeba culture medium was mixed with 20 μl of SuperFect transfection reagent (Qiagen), incubated for 10 min at room temperature, and then added to the cells. After incubation at 25°C for 3 hr to allow for uptake of the DNA-SuperFect complexes, the cells were washed with PBS, incubated for 24 hr, and placed in encystation medium and incubated for 24, 48, or 72 hr [
27]. The AcCP-EGFP expression in the amoeba at each time point was assessed by fluorescence microscopy. The cells were also counter-stained with LysoTracker Red DND 99 (Molecular Probes, Eugene, Oregon, USA), which labels acidic compartments such as lysosomes and autophagolysosomes, and the fluorescence was measured by using band-pass filters that provided excitation and emission wavelengths of 500 to 530 nm and 570 to 590 nm, respectively.
To further analyze expression pattern of AcCP in
A. castellanii, western blot analysis for different stages of the amoeba was performed. Encystation of trophozoites was induced by the method described above [
21,
22]. The amoeba cells were collected at 24, 48, or 72 hr after encystation induction. Lysates from
A. castellanii trophozoites and cysts collected at each time point were separated by SDS-PAGE. The proteins were transferred to polyvinylidene fluoride (PVDF) membranes, which were then blocked overnight with 5% skim milk in Tris-buffered saline (TBS) at 4°C, washed with TBS containing 0.05% Tween 20 (TBST), and incubated with anti-AcCP (1:1,000) for 2 hr at room temperature. After washing with TBST several times, the membranes were incubated with anti-mouse horseradish peroxidase-labeled IgG (1:2,000; Dako, Glostrup, Denmark) for 30 min at room temperature. Proteins were detected with enhanced chemiluminescence (ECL) western blotting detection reagents (Amersham Biosciences, Piscataway, New Jersey, USA). Membranes were stripped and probed with a mouse monoclonal anti-actin antibody (1:5,000; Santa Cruz Biotechnology, Santa Cruz, California, USA) for 2 hr to detect
Acanthamoeba actin as a loading control [
27]. Membranes were washed with TBST and incubated with anti-mouse horseradish peroxidase-labeled IgG (1:2,000; Dako) for 30 min at room temperature and the proteins were detected with ECL reagents (Amersham Biosciences). Western blot analysis for cultured medium collected at 24, 48, or 72 hr incubation of trophozites was also performed with anti-AcCP to analyze secretion of AcCP to culture medium as the same method described above.
RESULTS
Identification of a gene encoding
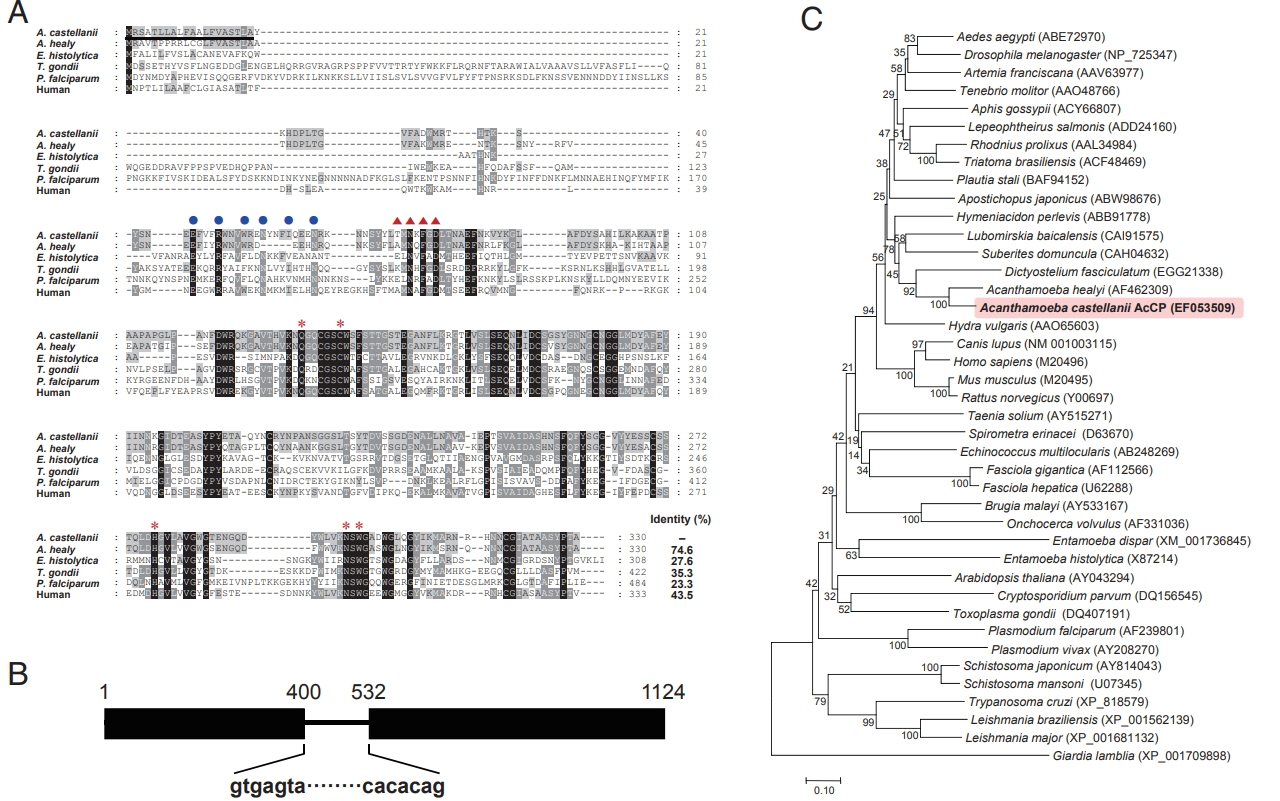
A. castellanii cathepsin L-like cysteine protease (AcCP) Following PCR with degenerate primers and subsequent sequencing analysis of 20 randomly selected clones, a partial gene sequence encoding AcCP, which clearly distinguished from all presently known
Acanthamoeba cysteine proteases, was obtained. The sequence encoding full-length AcCP gene was finally obtained using RACE procedures. The AcCP gene (Gen-Bank accession number: EF053509) had a 993-bp open reading frame encoding a protein of 330 amino acids, with a predicted molecular weight of 36.3 kDa. Multiple sequence alignment of the primary amino acid sequence of AcCP with those of related cathepsin-L family enzymes from several parasites and human revealed sequence identity ranged from 23.3% to 74.6% (
Fig. 1A). The 5 amino acid residues (Q, C, H, N, and W) that form the catalytic site, which are essential for stabilization of either the thiolate-imidazolium ion pair or the transition state, of cathepsin L-family cysteine proteases are well conserved in AcCP. The modified ERFNIN and GNFD motifs that are conserved in cathepsin L-like cysteine protease family members were also found in AcCP. Predicted signal peptide sequence was identified in the N-terminal portion of AcCP. When analyzed genomic DNA structure of AcCP by comparing the genomic and cDNA sequences, it was found that the gene was interrupted by an intron with the size of 131 bp (
Fig. 1B). Phylogenetic analysis of AcCP with cathepsin L cysteine proteases from other organisms revealed that AcCP was clustered into a clade with cysteine proteases from
A. healyi (Gen-Bank accession No. AF462309) and
Dictyostelium fasciculatum (GenBank accession No. EGG21338) (
Fig. 1C).
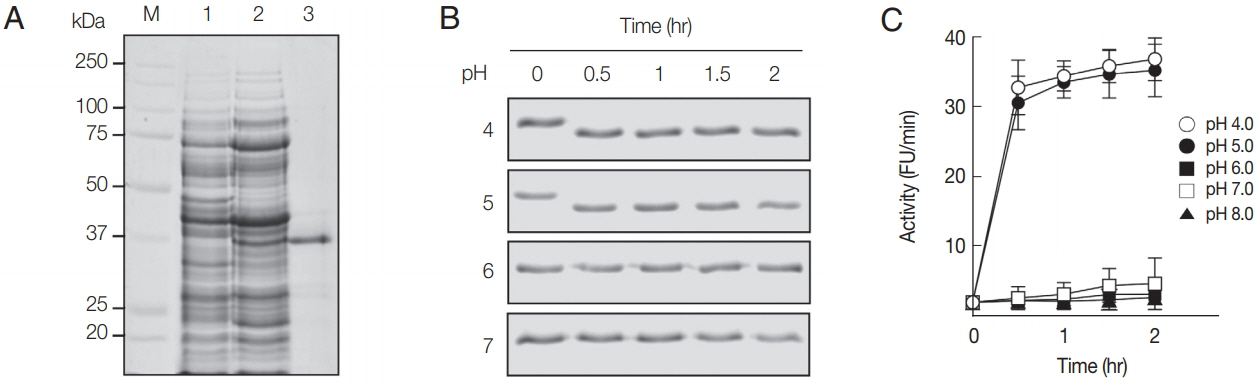
The rAcCP, comprising a portion of the prodomain and entire mature domain, was expressed in
E. coli as an insoluble protein, with an approximate molecular weight of 36 kDa, which is coincided with estimated molecular mass from the deduced amino acid sequences (
Fig. 2A). The purified rAcCP was refolded under alkaline conditions and further incubated in different pHs to allow auto-catalytic processing. The refolded rAcCP was processed to the enzymatically-active species of approximately 30 kDa under acidic conditions, pH 4.0 and 5.0 (
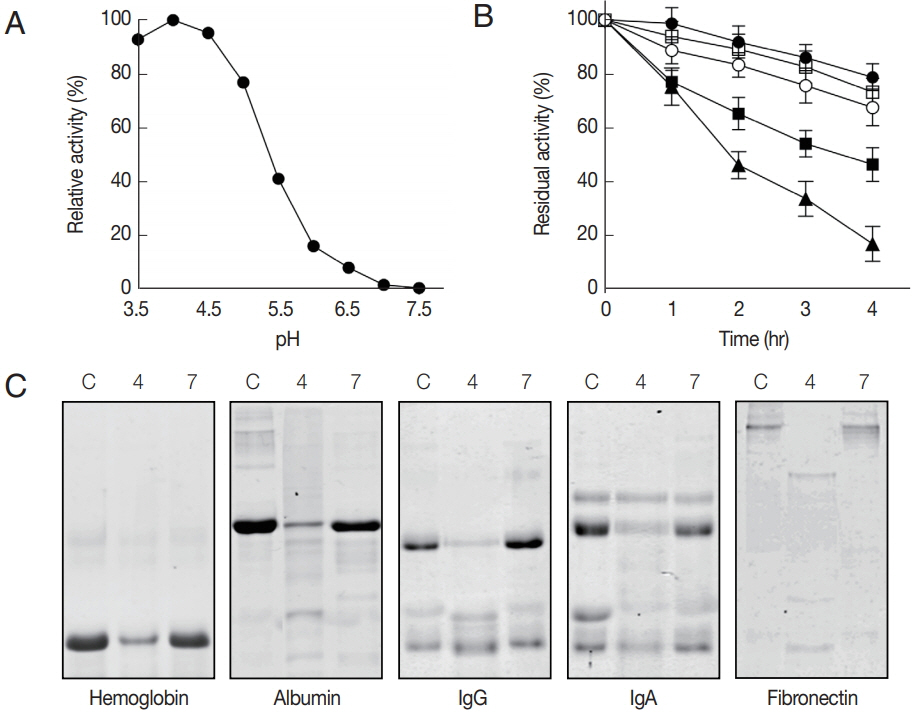
Fig. 2B, C). The fully processed rAcCP was effectively inhibited by a cysteine protease inhibitor, E-64 (data not shown). The fully activated rAcCP showed hydrolytic activity in acidic conditions with an optimum at pH 4.0 (
Fig. 3A). The enzyme was relatively stable at neutral pH values, but it was unstable under acidic pHs (
Fig. 3B). The hydrolytic activity of rAcCP for several human proteins, including hemoglobin, albumin, IgG, IgA and fibronectin, was analyzed. The recombinant enzyme effectively hydrolyzed all tested proteins under acidic conditions (pH 4.0), but did not substantially degrade the proteins under neutral conditions (pH 7.0) (
Fig. 3C). Only fibronectin was partially hydrolyzed by rAcCP at neutral pH. The substrate specificity of rAcCP was characterized with several fluorogenic dipeptidyl substrates. The enzyme showed strong activity against Z-FR-MCA and Z-LR-MCA, but did not against Z-RRMCA (
Table 1).
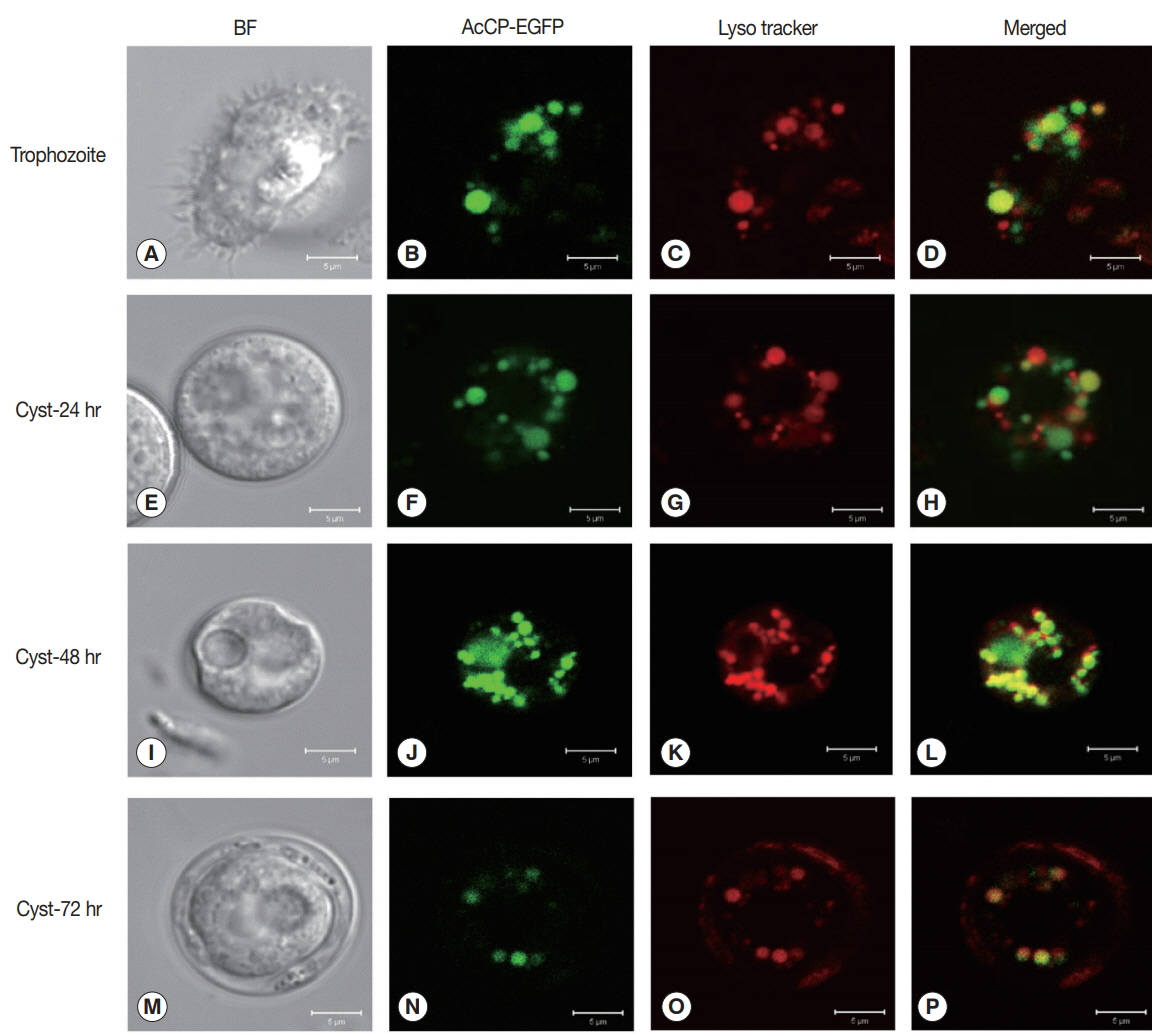
Cellular localization of AcCP was determined using an
Acanthamoeba vector expressing AcCP with a C-terminal EGFP fusion (AcCP-EGFP).
A. castellanii trophozoites were transfected with the plasmid and then examined by fluorescence microscopy followed by counter staining with LysoTracker Red. The AcCP-EGFP fusion protein was distributed in multiple vesicle-like structures in the cytoplasm of both trophozoites and cysts, which were colocalized with the LysoTracker-labeled compartments, indicating that AcCP was localized in lysosomes (
Fig. 4). Western blot analysis using anti-AcCP was performed to evaluate the expression profile of AcCP in different developmental stages of amoeba, trophozoites and cysts. The results indicated that AcCP expression was observed in both trophozoites and cysts with a maximum at trophozoite stage (
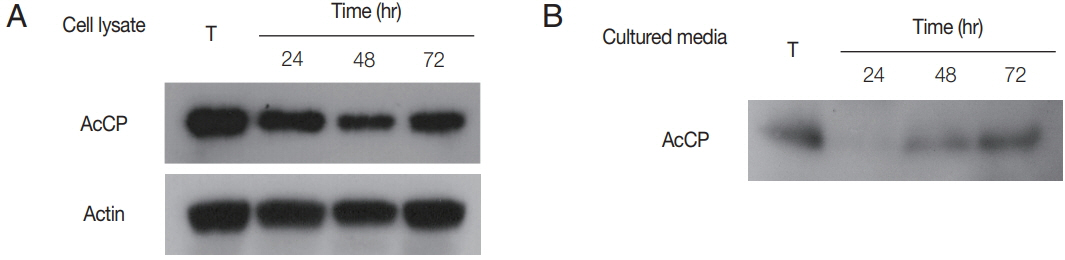
Fig. 5). Western blot analysis for cultured medium revealed that AcCP was detected in cultured medium of trophozoites (
Fig. 5). These results collectively suggested that AcCP was expressed in all developmental stages of the
Acanthamoeba and was secreted or released from the trophozoites.
DISCUSSION
In this study, we isolated a gene encoding AcCP and characterized its biochemical and biological properties. Sequence analysis of AcCP suggests that this enzyme is a typical cathepsin L family cysteine protease, which shares similar structural characteristics with other cathepsin L-like enzymes. The ERFNIN and GNFD motifs, which are well-conserved and play an important role in the processing and folding of cathepsin L family cysteine proteases [
28,
29], are well conserved in the pre-prodomain of AcCP. The amino acid residues (Q, C, H, N, and W) that form the catalytic site of cathepsin L family cysteine proteases are also well conserved in AcCP. Optimum hydrolytic activity at acidic pHs, requirement of reduced condition for maximum activity and specific inhibition by E-64 also suggest the enzyme is a typical cathepsin L family enzyme.
The optimum pH for rAcCP in acidic conditions suggests that the enzyme is suited for activity in acidic conditions. Continuous expression of AcCP during the developmental stages from trophozoites to cysts suggests its essential roles in the survival and development of
A. castellanii. Considering its cellular localization in the lysosomal compartments and broad hydrolytic activity against various proteins including hemoglobin, albumin, IgG, IgA and fibronectin at acidic pH, the primary biological function of the enzyme seems to be related to host protein digestion engulfed by the amoeba. The finding that AcCP is secreted or released into the culture medium of
A. castellanii trophozoites also suggests another possible biological role for the enzyme. Fibronectin, one of main components of the extracellular matrix of epithelial cells, hydrolysis activity of AcCP at neural pH, pH 7.0, suggests its potential role in tissue invasion by the amoeba. High level of AcCP expression during encystation stage also postulates another potential biological function of the enzyme. The potential roles for cysteine proteases in encystation or excystation have been suggested in a number of protozoan parasites, such as
Sterkiella historiomuscorum [
30],
Giardia lamblia [
31–
33],
Giardia muris [
34],
Entamoeba invadens [
35–
38], and
A. castellanii [
16,
17]. In this study, we did not obtain direct evidence for involvement of AcCP in encystations or excystation of
A. castellanii. However, considering to its continuous expression in the stages during encystation of
A. castellanii, AcCP may play a role in the encystation of the amoeba, but this should be elucidated further.
In conclusion, we identified and characterized a novel cathepsin L family cysteine protease, AcCP, in A. castellanii. Considering to lysosomal localization, secretion or release by trophozoites and continuous expression in trophozoites and cysts, the enzyme could be a multifunctional enzyme that plays important biological functions for nutrition, development and pathogenicity of A. castellanii. These results also imply that AcCP can be a promising target for development of chemotherapeutic drug for Acanthamoeba infections. Although, partial biochemical and functional characterization of Acanthamoeba cysteine proteases have been done, the biological roles of cysteine proteases of Acanthamoeba have not been fully understood yet and further comprehensive studies are needed for a more in-depth understanding of the biological significance of these enzymes.
Notes
-
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGMENT
This Work was supported by Development Fund Foundation, Gyeongsang National University, 2015.
Fig. 1Sequence alignment and phylogenetic analysis. (A) Multiple sequence alignment of AcCP (EF053509) with the cysteine proteases of other protozoan parasites and human including Acanthamoeba healyi (AF462309), Entamoeba histolytica (X87214), Toxoplasma gondii (XP_002371694), Plasmodium falciparum (XP_001347836) and human (M20498). The dashes represent gaps introduced to maximize alignment. The ERFNIN motif, which is well conserved in the cathepsin L-like subgroup, was marked with closed blue circles. The GNFD motif was shown as red triangles. The predicted signal peptide region was shown with a black line. Red asterisks indicate the conserved active site residues. (B) Genomic DNA structure. AcCP gene was interrupted by an intron with the size of 131 bp in genomic DNA sequence. (C) Phylogenetic analysis. The tree was built with the neighbor-joining method using the MEGA4 program. Numbers on the branches indicate bootstrap proportions (1,000 replicates). Full-length amino acid sequences for each gene were analyzed.
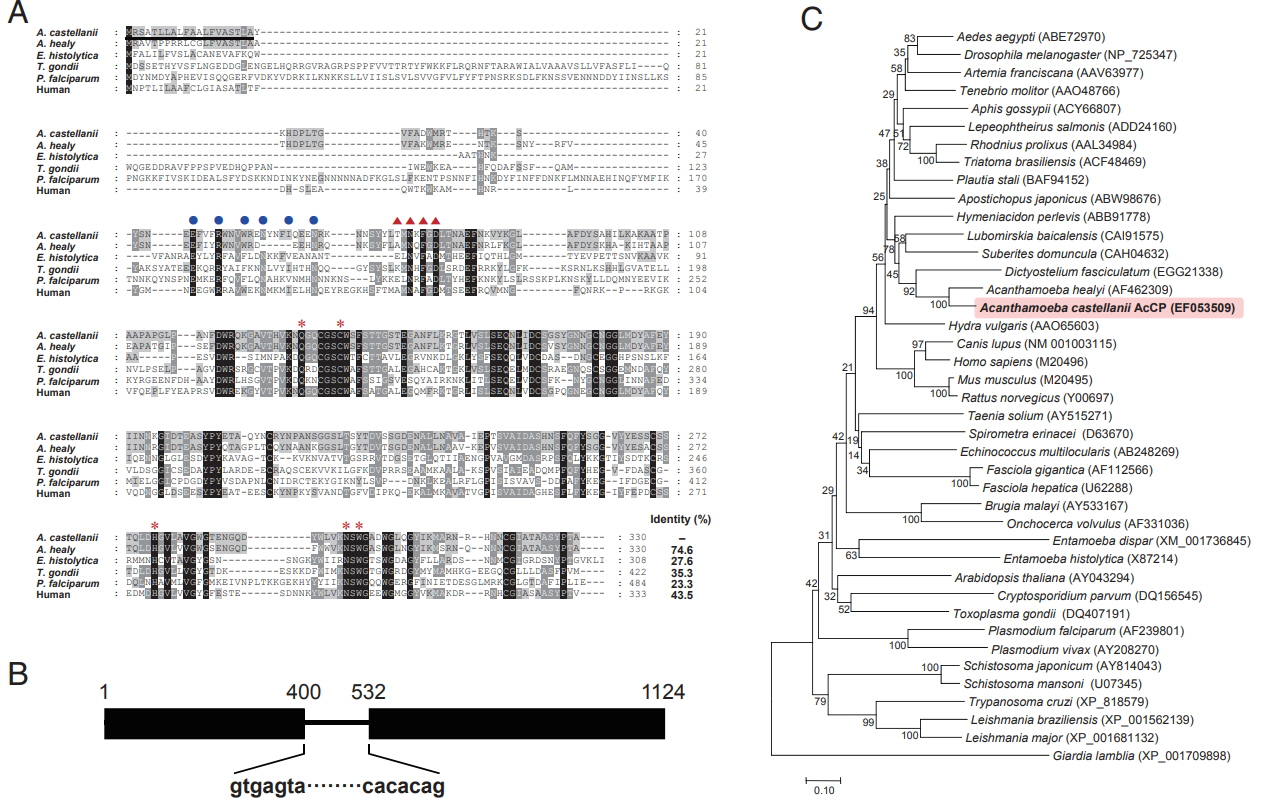
Fig. 2Production and refolding of rAcCP. (A) Expression and production of rAcCP. Proteins were analyzed by SDS-PAGE and stained with Coomassie blue. Lane 1, Escherichia coli lysate control (30 μg); lane 2, isopropyl-1-thio-β-D-galactopyranoside-induced E. coli lysate (30 μg); lane 3, rAcCP purified by Ni–NTA affinity chromatography (5 μg). Lane M, molecular size marker proteins. (B) Autocatalytic processing of refolded rAcCP. The purified rAcCP was refolded and incubated at different pHs. Aliquots were collected at indicated times and were analyzed by 12% SDS-PAGE with Coomassie staining. (C) Enzyme activity assay. The refolded rAcCP was incubated in different pH buffers and enzyme activity was assayed with Z-FR-MCA as a substrate.
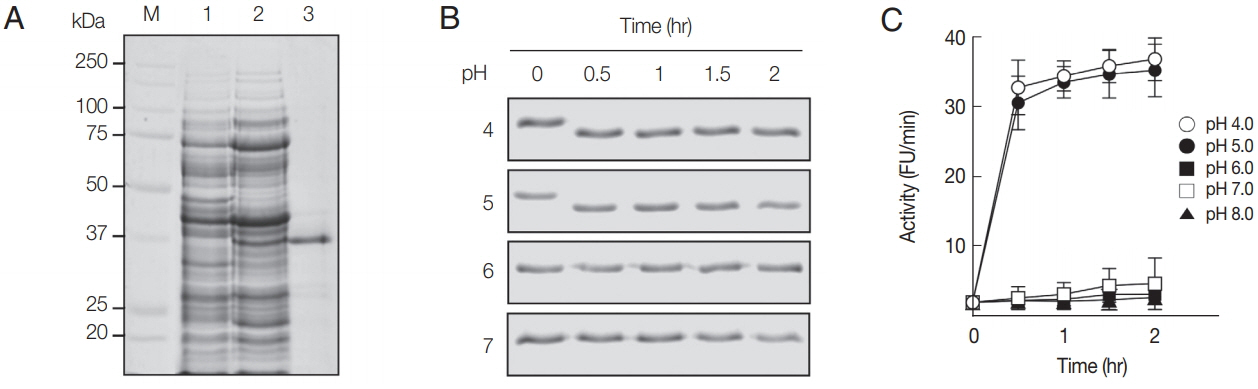
Fig. 3Characterization of biochemical properties of rAcCP. Biochemical properties of fully activated rAcCP were analyzed. (A) Optimal pH. Enzyme activity was assayed in various pH buffers ranging from pH 3.5–7.5. Maximal activity was shown as 100%. (B) Enzyme stability. rAcCP was incubated in various pH buffers at 37°C for the indicated time and residual enzyme activity was assayed. All the assays were performed with Z-LR-MCA as a substrate. pH 4.0 (▲), pH 5.0 (■), pH 6.0 (□), pH 7.0 (●), and pH 8.0 (○). (C) Degradation of human proteins by rAcCP. Each macromolecular protein substrate was incubated with rAcCP (100 nM) in 50 mM sodium acetate (pH 4.0) or 50 mM sodium phosphate (pH 7.0) in the presence of 1 mM GSH for 3 hr at 37°C and analyzed by SDS-PAGE. Lane C, protein without rAcCP; lanes 4 and 7, incubated with rAcCP at pH 4.0 and pH 7.0, respectively.

Fig. 4Cellular localization of AcCP. Trophozoites of A. castellanii were transfected with pGAPDH-AcCP-EGFP to analyze locations of AcCP-EGFP expression in the amoeba. The transfected A. castellanii trophozoites (A–D) were transferred into encystation medium, incubated for 24 hr (E–H), 48 hr (I–L), and 72 hr (M–P), and then examined under a fluorescence microscope (green). Lysosomes and autophagolysosomes were visualized by counter-staining with LysoTracker Red (red), and the resulting images were merged (yellow). BF denotes bright-field images. Scale bars indicate 5 μm.
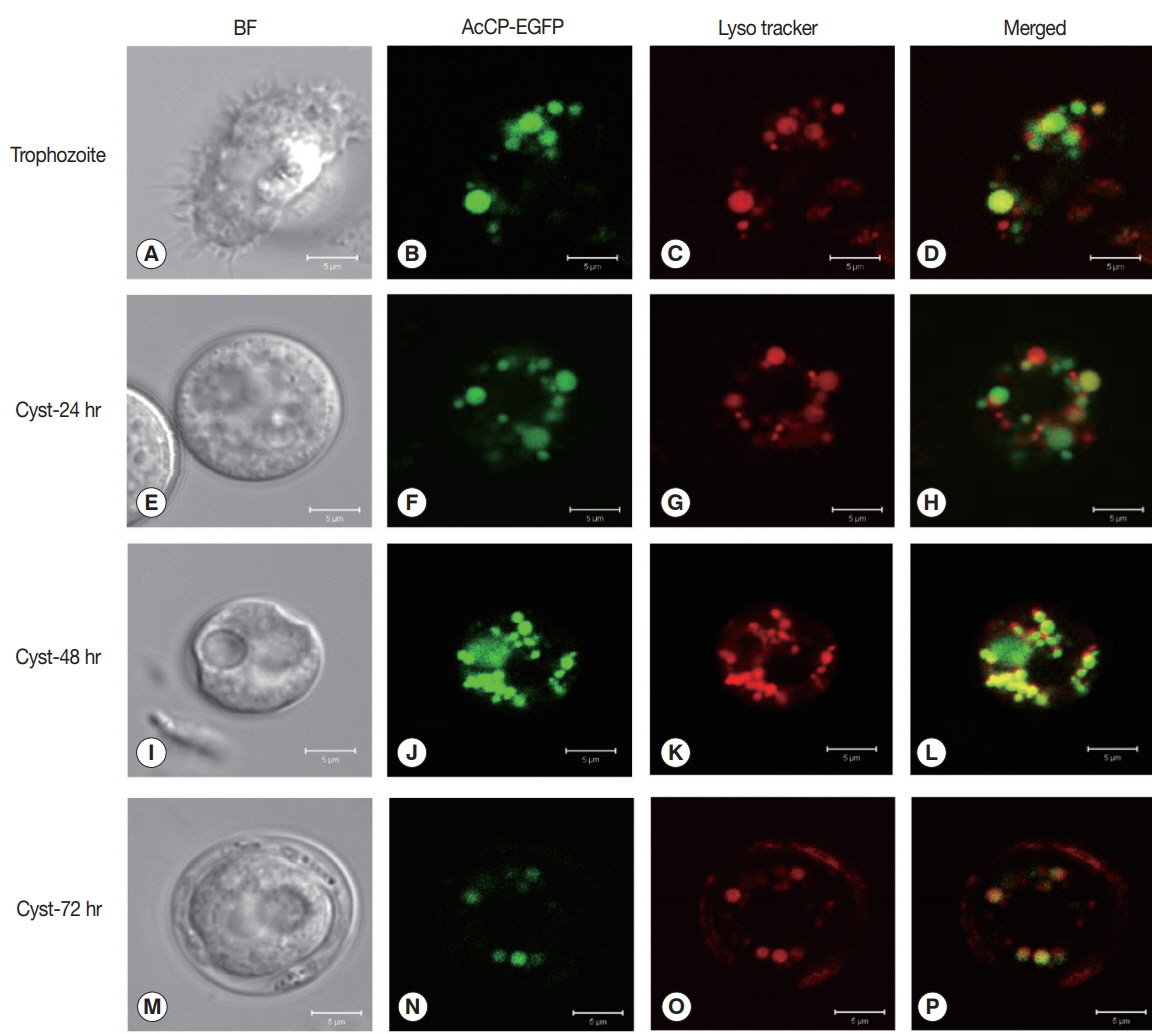
Fig. 5Expression profiles of AcCP. (A) Expression profile of AcCP in different developmental stages of A. castellanii. A. castellanii trophozoites were encysted in encystation medium for 24, 48, or 72 hr. Lysates of A. castellanii trophozoites and cysts collected at each time point were separated by SDS-PAGE, transferred to PVDF membrane and probed with anti-AcCP and anti-actin. (B) Secretion of AcCP. The trophozoites were incubated in PYG medium at 25°C for 24, 48, or 72 hr and the cultured medium were collected at indicated time, respectively. Western blot analysis for cultured medium was performed with anti-AcCP. T, lysates of A. castellanii trophozoites.
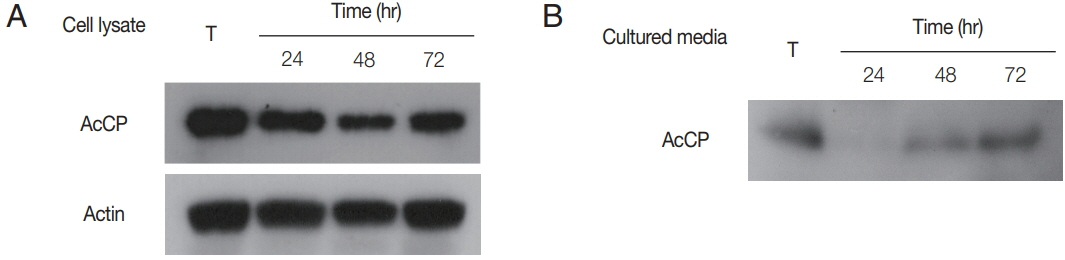
Table 1Kinetic parameters for AcCP
Table 1
|
Substrate |
kcat/Km (M−1s−1) |
|
Z-FR-MCA |
6.2×103
|
|
Z-LR-MCA |
2.1×104
|
|
Z-RR-MCA |
NH |
References
- 1. Khan NA, Siddiqui R.
Acanthamoeba affects the integrity of human brain microvascular endothelial cells and degrades the tight junction proteins. Int J Parasitol 2009;39:1611-1616.
- 2. Marciano-Cabral F, Cabral G.
Acanthamoeba spp. as agents of disease in humans. Clin Microbiol Rev 2003;16:273-307.
- 3. Bowers B. Comparison of pinocytosis and phagocytosis in Acan thamoeba castellanii
. Exp Cell Res 1977;110:409-417.
- 4. Bowers B, Olszewski TE.
Acanthamoeba discriminates internally between digestible and indigestible particles. J Cell Biol 1983;97:317-322.
- 5. Lloyd D, Turner NA, Khunkitti W, Hann AC, Furr JR, Russell AD. Encystation in Acanthamoeba castellanii: development of biocide resistance. J Eukaryot Microbiol 2001;48:11-16.
- 6. Khan NA. Acanthamoeba: Biology and Pathogenesis. Norfolk, UK. Caster Academic Press; 2009.
- 7. Weisman RA. Differentiation in Acanthamoeba castellanii
. Annu Rev Microbiol 1976;30:189-219.
- 8. Sajid M, McKerrow JH. Cysteine proteases of parasitic organisms. Mol Biochem Parasitol 2002;120:1-21.
- 9. Atkinson HJ, Babbitt PC, Sajid M. The global cysteine peptidase landscape in parasites. Trends Parasitol 2009;25:573-581.
- 10. Na BK, Kang JM, Cheun HI, Cho SH, Moon SU, Kim TS, Sohn WM. Cryptopain-1, a cysteine protease of Cryptosporidium parvum, does not require the pro-domain for folding. Parasitology 2009;136:149-157.
- 11. Kang JM, Bahk YY, Cho PY, Hong SJ, Kim TS, Sohn WM, Na BK. A family of cathepsin F cysteine proteases of Clonorchis sinensis is the major secreted proteins that are expressed in the intestine of the parasite. Mol Biochem Parasitol 2010;170:7-16.
- 12. Alfieri SC, Correia CE, Motegi SA, Pral EM. Proteinase activities in total extracts and in medium conditioned by Acanthamoeba polyphaga trophozoites. J Parasitol 2000;86:220-227.
- 13. de Serrano-Luna JJ, Cervantes-Sandoval I, Calderon J, Navarro-García F, Tsutsumi V, Shibayama M. Protease activities of Acanthamoeba polyphaga and Acanthamoeba castellanii
. Can J Microbiol 2006;52:16-23.
- 14. Hadas E, Mazur T. Proteolytic enzymes of pathogenic and non-pathogenic strains of Acanthamoeba spp. Trop Med Parasitol 1993;44:197-200.
- 15. Mitro K, Bhagavathiammai A, Zhou OM, Bobbett G, McKerrow JH, Chokshi R, Chokshi B, James ER. Partial characterization of the proteolytic secretions of Acanthamoeba polyphaga
. Exp Parasitol 1994;78:377-385.
- 16. Leitsch D, Köhsler M, Marchetti-Deschmann M, Deutsch A, Allmaier G, Duchêne M, Walochnik J. Major role for cysteine proteases during the early phase of Acanthamoeba castellanii encystment. Eukaryot Cell 2010;9:611-618.
- 17. Moon EK, Hong Y, Chung DI, Kong HH. Cysteine protease involving in autophagosomal degradation of mitochondria during encystation of Acanthamoeba
. Mol Biochem Parasitol 2012;185:121-126.
- 18. Ramírez-Rico G, Martínez-Castillo M, de la Garza M, Shibayama M, Serrano-Luna J.
Acanthamoeba castellanii proteases are capable of degrading iron-binding proteins as a possible mechanism of pathogenicity. J Eukaryot Microbiol 2015;62:614-622.
- 19. Yun HC, Kim KY, Park SY, Park SK, Park H, Hwang UW, Hong KM, Ryu JS, Min DY. Cloning of a cysteine proteinase gene from Acanthamoeba culbertsoni
. Mol Cells 1999;9:491-496.
- 20. Hong YC, Hwang MY, Yun HC, Yu HS, Kong HH, Yong TS, Chung DI. Isolation and characterization of a cDNA encoding a mammalian cathepsin L-like cysteine proteinase from Acanthamoeba healyi
. Korean J Parasitol 2002;40:17-24.
- 21. Bowers B, Korn ED. The fine structure of Acanthamoeba castellanii (Neff strain). II. Encystment. J Cell Biol 1969;41:786-805.
- 22. Picazarri K, Nakada-Tsukui K, Nozaki T. Autophagy during proliferation and encystation in the protozoan parasite Entamoeba invadens
. Infect Immun 2008;76:278-288.
- 23. Na BK, Kang JM, Sohn WM. CsCF-6, a novel cathepsin F-like cysteine protease for nutrient uptake of Clonorchis sinensis
. Int J Parasitol 2008;38:493-502.
- 24. Na BK, Bae YA, Zo YG, Choe Y, Kim SH, Desai PV, Avery MA, Craik CS, Kim TS, Rosenthal PJ, Kong Y. Biochemical properties of a novel cysteine protease of Plasmodium vivax, vivapain-4. PLoS Negl Trop Dis 2010;4:e849.
- 25. Lee JY, Song SM, Moon EK, Lee YR, Jha BK, Danne DB, Cha HJ, Yu HS, Kong HH, Chung DI, Hong Y. Cysteine protease inhibitor (AcStefin) is required for complete cyst formation of Acanthamoeba
. Eukaryot Cell 2013;12:567-574.
- 26. Bateman E. Expression plasmids and production of EGFP in stably transfected Acanthamoeba
. Protein Expr Purif 2010;70:95-100.
- 27. Moon EK, Chung DI, Hong YC, Kong HH. Characterization of a serine proteinase mediating encystation of Acanthamoeba
. Eukaryot Cell 2008;7:1513-1517.
- 28. Vernet T, Berti PJ, de Montigny C, Musil R, Tessier DC, Ménard R, Magny MC, Storer AC, Thomas DY. Processing of the papain precursor. The ionization state of a conserved amino acid motif within the Pro region participates in the regulation of intramolecular processing. J Biol Chem 1995;270:10838-10846.
- 29. Shinde U, Inouye M. Intramolecular chaperones: polypeptide extensions that modulate protein folding. Semin Cell Dev Biol 2000;11:35-44.
- 30. Villalobo E, Moch C, Fryd-Versavel G, Fleury-Aubusson A, Morin L. Cysteine proteases and cell differentiation: excystment of the ciliated protist Sterkiella histriomuscorum
. Eukaryot Cell 2003;2:1234-1245.
- 31. Touz MC, Nores MJ, Slavin I, Carmona C, Conrad JT, Mowatt MR, Nash TE, Coronel CE, Luján HD. The activity of a developmentally regulated cysteine proteinase is required for cyst wall formation in the primitive eukaryote Giardia lamblia
. J Biol Chem 2002;277:8474-8481.
- 32. Touz MC, Lujan HD, Hayes SF, Nash TE. Sorting of encystation-specific cysteine protease to lysosome-like peripheral vacuoles in Giardia lamblia requires a conserved tyrosine-based motif. J Biol Chem 2003;278:6420-6426.
- 33. DuBois KN, Abodeely M, Sakanari J, Craik CS, Lee M, McKerrow JH, Sajid M. Identification of the major cysteine protease of Giardia and its role in encystation. J Biol Chem 2008;283:18024-18031.
- 34. Ward W, Alvarado L, Rawlings ND, Engel JC, Franklin C, McKerrow JH. A primitive enzyme for a primitive cell: the protease required for excystation of Giardia
. Cell 1997;89:437-444.
- 35. Sharma M, Hirata K, Herdman S, Reed S.
Entamoeba invadens: characterization of cysteine proteinases. Exp Parasitol 1996;84:84-91.
- 36. Gonzalez J, Bai G, Frevert U, Corey EJ, Eichinger D. Proteasome-dependent cyst formation and stage-specific ubiquitin mRNA accumulation in Entamoeba invadens
. Eur J Biochem 1999;264:897-904.
- 37. Makioka A, Kumagai M, Kobayashi S, Takeuchi T.
Entamoeba invadens: cysteine protease inhibitors block excystation and metacystic development. Exp Parasitol 2005;109:27-32.
- 38. Ebert F, Bachmann A, Nakada-Tsukui K, Hennings I, Drescher B, Nozaki T, Tannich E, Bruchhaus I. An Entamoeba cysteine peptidase specifically expressed during encystation. Parasitol Int 2008;57:521-524.