Distribution Status of Hybrid Types in Large Liver Flukes, Fasciola Species (Digenea: Fasciolidae), from Ruminants and Humans in Vietnam
Article information
Abstract
The aim of this study is to delineate ‘admixed hybrid’ and ‘introgressive’ Fasciola genotypes present in the Fasciola population in Vietnam. Adult liver flukes collected from ruminants in 18 Provinces were morphologically sorted out by naked eyes for small (S), medium (M) and large (L) body shapes; and human samples (n=14) from patients. Nuclear ribosomal (rDNA) ITS1 and ITS2, and mitochondrial (mtDNA) nad1 markers were used for determination of their genetic status. Total 4,725 worm samples of ruminants were tentatively classified by their size: 6% (n=284) small (S)-, 13% (n=614) medium (M)-, and 81% (n=3,827) large (L)-forms. All the representative (n=120, as 40 each group) and 14 human specimens, possessed maternal mtDNA of only F. gigantica and none of F. hepatica. Paternally, all (100%) of the L-(n=40) and 77.5% (n=31) of the M-flukes had single F. gigantica rDNA indicating ‘pure’ F. gigantica. A majority (90%, n=36) of the S- and 15% (n=6) of the M-worms had single F. hepatica rDNA, indicating their introgressive; the rest (10%, n=4) of the S- and 7.5% (n=3) of the M-flukes had mixture of both F. gigantica and F. hepatica rDNAs, confirming their admixed hybrid genetic status. Fourteen human samples revealed 9 (64%) of pure F. gigantica, 3 (22%) of introgressive and 2 (14%) of admixed hybrid Fasciola spp. By the present study, it was confirmed that the small worms, which are morphologically identical with F. hepatica, are admixed and/or introgressive hybrids of Fasciola spp., and able to be the pathogens of human fascioliasis.
INTRODUCTION
Fasciola species (Digenea: Fasciolidae) are the large liver flukes, which primarily infect ruminants and accidentally humans, and then particularly has the socio-economic importance as the food-borne zoonotic trematodes [1]. F. hepatica and F. gigantica are the two taxonomically recognized species, where the former is distributed in temperate zone and the latter mainly in sub- and tropical countries. Fertilizations have been reported as of their typical reproductive features, within and between these two Fasciola species [2]. The co-existence of both species and the ‘intermediate’ form comprising admixed/introgressive hybrids of Fasciola spp., have been documented in numerous countries [1,3–8]. The intermediate form of both hybrid genotypes, up-to-date, has been reported present in various Asian and African countries, including Vietnam, China, Korea, Japan, Thailand, Iran, Bangladesh, Pakistan, Nepal, India, Myanmar, and Egypt. Morphologically, many of them look like F. hepatica, but genetically, their genome is mixed between F. hepatica and F. gigantica [3,6,7,9–18]. In the particular, if morphologically examined, F. gigantica is a long ‘leaf shape’ fluke with the physiometrical ratio between the body length (BL) and body width (BW) is about 4–5/1; and F. hepatica is a short ‘leaf shape’ with prominent shoulder, of about 2–3/1 BL/BW ratio; and no standard metrics applied for the ‘hybrid’ species [7,19,20].
If merely based on physiometrics and morphology, the intermediate form of Fasciola is easy to be confused with the real F. hepatica species. Hence, up-to-date, there have been various molecular approaches/assays developed for identification and discrimination of these species/genotypes including PCR-RFLP [10,21]; PCR-SSCP [3]; loop-mediated isothermal amplification (LAMP) [22,23]; and PCR-coupled sequence analysis [11,14,24–29]. Recently, the effective approaches are employed for detecting the hybrid Fasciola flukes using nuclear protein coding genes, (ie., pepck and pold) [17,18,25] and intron regions of the taurocyamine kinase gene [8]. The mostly used discriminative markers have been relying on polymorphic nucleotide sites in nuclear ribosomal ITS1 and ITS2, and in mitochondrial genes, nad1 and cox1 or else, and commonly applied in many studies to date [3,5,7,10,11,13,14,16,20,21,24].
In Vietnam, fascioliasis is highly endemic, posing heavy burden in ruminants and humans, particularly in Central Provinces; and the interspecific hybridization between F. hepatica and F. gigantica has been nationwide reported [11,12,14,24,30,31]. Such a tropical country like Vietnam, where live F. hepatica does not exist, and only F. gigantica present, how these hybridized genotypes have been formed, it is still a questionable issue to be clarified [1,11,12,14,24,30]. Identical F. hepatica-shaped flukes have been found with a large number in ruminants, and commonly referred to as intermediate forms of Fasciola spp. Studies on the molecular delineation of the genetic status of 2 genotypes of hybridization: which exactly admixed hybrid or introgressive Fasciola spp. present in the infective Fasciola population in Vietnam, appeared to be limited [11,12,24,30].
The present study was conducted to determine the physiometrical shapes of Fasciola spp. in ruminants by on-site examination of flukes collected from slaughtered ruminants (buffaloes, cattle, sheep, goats) and the genetic delineation of admixed hybrid and introgressive genotypes by analysis of PCR–based sequencing of the nuclear ribosomal internal transcribed spacer 1 and 2 (ITS1 and ITS2) and the mitochondrial nicotinamide dehydrogenase subunit 1 (nad1) gene. In addition, samples from human cases were also molecularly clarified for genotypic hybridization. Data in this study provide understandings of to what extent these special hybridized genotypes present in the Fasciola population in Vietnam.
MATERIALS AND METHODS
Sample collection
During 10 years (2007–2016), total 4,725 adult worms were collected from the livers of slaughtered ruminants, by provincial veterinary technicians from 11 Provinces in the North: 1. Cao Bang (95 flukes), 2. Lang Son (121), 3. Bac Kan (56), 4. Yen Bai (98), 5. Thai Nguyen (2,170), 6. Ha Noi (772), 7. Hung Yen (135), 8. Hai Duong (88), 9. Ninh Binh (218), 10. Nghe An (94), 11. Ha Tinh (66); and 7 Provinces in the Central of Vietnam: 12. Thua Thien-Hue (123), 13. Quang Nam (36), 14. Binh Dinh (125), 15. Phu Yen (104), 16. Khanh Hoa (188), 17. Dak Lak (79), 18. Ninh Thuan (157). Flukes were originally washed in physiological saline (0.9% NaCl), fixed mainly in 70% or 96% ethanol, or in formalin or freshly frozen. Human samples (n=14) were provided by the hospital physicians taken from patients from Ha Noi (6 samples), Nghe An (1), Quang Binh (1), Thua Thien-Hue (1), Quang Nam (1), Binh Dinh (3) and Khanh Hoa (1) Provinces (Table 1). Ethical approval was applied for human cases from the National Institute of Malariology, Parasitology and Entomology (NIMPE) on behalf of the Ministry of Health.
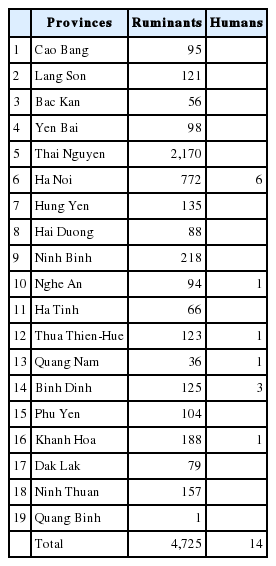
No. of Fasciola samples collected from Northern and Central Provinces in Vietnam from ruminants and humans
Groupings of worm samples
Worm samples preserved in formalin or ethanol or frozen, were examined with naked eyes and sorted out by their body size: small (S), medium (M) and large (L). The majority of samples from human cases were provided in pieces, unable to be examined by shape, thus, they were used directly to molecular analysis. Worms were taken per each group of L (n=40), M (n=40) and S (n=40), preferably those fixed in ethanol or frozen, and all samples from humans (n=14) to molecular analysis. PCR and sequencing were used to obtain nuclear ribosomal ITS1, ITS2 and mitochondrial nad1 for sequence analysis. Subsequently, based on these Fasciola-species specific markers, F. hepatica, F. gigantica and admixed hybrid or introgressive Fasciola spp. were clearly delineated for evaluation.
Genomic DNA extraction and PCR
Following rehydration of the worms, genomic DNA was extracted from individual worms using the GeneJETTM Genomic DNA Purification Kit (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA), according to the manufacturer’s instructions. Primers listed in Table 2 were used for PCR amplification of the entire ITS1 spanning ITS2 region (1,520 bp) using primer pair U18S2F/U28S2R or alternatively, of each, ITS1 (852 bp) by U18S2F/U3SR and ITS-2 by U3SF/U28S2R primers; and mitochondrial nad1 gene using primer pair FANDF/FANDR, respectively.

Primer sequences used for amplification and sequencing internal transcribed spacers (ITS) and mitochondrial nad1 gene for molecular discrimination of Fasciola species
DNA template (−100 ng DNA) or no DNA (negative control) in 2 μl was added to 48 μl of PCR reaction buffer containing 25 μl PCR Master Mix (Thermo Fisher Scientific), 2 μl of each primer (10 pmol/μl), 2 μl dimethyl sulfoxide (DMSO), 17 μl H2O. PCR was conducted at 94°C for 5 min, followed by 35 cycles of 94°C/30 sec, 52°C/30 sec (annealing), 72°C for 2 min (extension), followed 10 min/72°C (final extension). PCR products, stained with ethidium bromide, were visualized by electrophoresis in 1.0% agarose gels (Wealtec, Sparks, Nevada, USA). Following the purification using the GeneJET Gel Extraction Kit (Thermo Fisher Scientific), amplicons were directly sequenced using the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems, Foster City, California, USA) via a commercial service provider (Macrogen Inc., Seoul, Korea).
Analysis of nucleotide markers for species discrimination of Fasciola spp
From the previous studies [3,7,11,12,16,21,24], key species-markers for identification and discrimination of Fasciola spp. and admixed hybrid/introgressive genotypes have been clearly recognized.
For nuclear ribosomal markers, alignments were conducted over sequences obtained from ITS1 (421 bp) for finding 5 polymorphic sites, including the key RsaI cutsite (GTAC, at positions 296–299) in F. hepatica, and non-RsaI cutsite (GTAT) in F. gigantica [21]; and from ITS-2 (361–362 bp) for Thymine (T) nucleotide at position 327, present in F. hepatica and absent in F. gigantica [11].
Phylogenetic analysis and determination of Fasciola gigantica nad1-haplogroups
Mitochondrial haplogroups of the specimens were determined by reconstruction of phylogenetic tree based on the alignment analysis of nad1 (trimmed to 535 bp) with the reference sequences from F. hepatica, F. gigantica haplogroups of A, B, C, D, E and intermediate forms of Fasciola spp. from the GenBank database [7] (Table 3).

List of nad1 reference sequences and their accession numbers from GenBank and published database used for phylogenetic analysis of F. gigantica haplogroups in the present study
The alignments of nad1 sequences were performed using GENEDOC 2.7 (http://iubio.bio.indiana.edu/soft/molbio/ibmpc/genedoc-readme.html) and phylogenetic analysis by MEGA 7.0 (www.megasoftware.net), using genetic distance calculation by Kimura 2-parameter model and maximum likelihood method tested by bootstrapping with 1,000 replications [32]. The nad1 sequences representing haplogroups of A, B, C, D, E were selected for reference and included in the phylogenetic analysis.
RESULTS
Proportion of recovered worms by the groups
All 4,725 worms collected from slaughtered ruminants (buffaloes, cattle, sheep, goats), were individually examined with naked eyes and classified by their morphological features with the ratio of body length (BL) to body width (BW). They were divided into 3 groups by size of their appearance: small (S), about 2.0–3.6×1.3 cm; medium (M), 3.6–4.2×1.3 cm; and large (L) groups, 4.2–6.5×1.2 cm (Fig. 1; Table 4). The mean ratio of BL to BW, was able to be inferred for flukes, as: 1.54–2.77/1 (S); 2.77–3.23/1 (M), and 3.23–5.4/1 (L), respectively (Table 4). The number of worms was 284 (6.0%) in S-group, 614 (13.0%) in M-group and 3,827 (81.0%) in L-group (Table 4).

Representative adult individuals sorted out for groups of small (S), medium (M) and large (L) shaped Fasciola liver flukes based on morphological examination by their size and the inferred ratio (BL/BW) between the body length (BL) and body width (BW). Size is indicated by a bar (1.2 cm). (A) fresh; (B) preserved flukes.

No. of small (S), medium (M) and large (L) individual shapes based on morphological examination of flukes present in the Fasciola spp. population (n=4,725 adults) from ruminants
Genotypic discrimination based on nuclear ribosomal markers of ITS1 and ITS2
Ribosomal DNA region of 852 bp containing the complete ITS1 (421 bp), numbering according to [3] and ITS2 sequences (361–362 bp), by [11] were amplified and sequenced from 120 flukes of ruminants and 14 human samples. Alignment of the sequences showed 5 polymorphic sites in ITS1 and 1 most distinguishable position in ITS2. Table 5 showed that there were 3 genotypic sequences of rDNA obtained, at the positions 17-107-201-279-299 corresponding to ITS1, and position at 327 corresponding to ITS2: i) T-T-T-A-T and (-), referred to as of F. gigantica; ii) C-A-C-T-C and T referred to as of F. hepatica; and iii) Y-W-Y-W-Y and (T/-), a mixture of the above 2 sequences, indicative of the presence of F. hepatica and F. gigantica in an individual. At positions 299–296 in ITS1, GTAC sequence of a RsaI cutsite is permanently present in F. hepatica (termed as RsaI(+)); and GTAT (non-RsaI) in F. gigantica (RsaI(−)); and at position 327 in ITS2, F. hepatica possesses a T nucleotide while F. gigantica does not (Table 5).

Molecular delineation of Fasciola species and types of hybridization among S-, M- and L-shaped liver flukes from ruminants and human samples in Vietnam based on the key nucleotide markers of ITS1 and ITS2 and mitochondrial nad1
The RsaI(+/−) in ITS1 and (T/-) in ITS2 were the key markers used to delineate the genetic status in the samples sequenced. All (100%) of the L-shaped (n=40) and 77.5% (n=31) of the M-shaped flukes of ruminants had F. gigantica rDNA indicating that they are ‘pure’ F. gigantica. A major portion (90%, n=36) of the S-shaped and 15% (n=6) of the M-shaped flukes had single F. hepatica rDNA, indicating them introgressive. The rest (10%, n=4) of the S-shaped, and 7.5% (n=3) of the M-shaped individuals had a mixture of both, F. gigantica and F. hepatica rDNAs, confirming their admixed hybrid genetic status. Of 120 fluke samples from ruminants, 71 were F. gigantica, 42 were introgressive and 7 were admixed hybrid Fasciola spp. (Table 5).
Fourteen samples (n=14) from humans revealed 9 (64%) of pure F. gigantica, 3 (22%) of introgressive and 2 (14%) of admixed hybrid Fasciola spp. (Table 5).
Mitochondrial haplogroups based on nad1 sequences
Fragment of 1,170 bp covering mitochondrial nad1 (903 bp) was obtained from 45 introgressive (42 ruminant and 3 human samples), 9 “admixed” (7 ruminant and 2 human samples) Fasciola sp., and 80 F. gigantica samples (71 from ruminants and 9 from humans) (Table 5). Alignment analysis of a portion of nad1 (trimmed to 535 bp) indicated notable diversity among 3 genotypes of Fasciola spp. The Vietnamese Fasciola spp. generated 2 additional haplogroups, designated as VN1 and VN2. All the admixed and introgressive sequences from 54 samples from this study together with those intermediate Fasciola spp. of Vietnam, China, Myanmar, Korea, Japan, constituting VN1 group in a sister position with respect to haplogroup A. Another VN2 is a cluster comprising all nad1 sequences from ‘pure’ F. gigantica species from China and Vietnam (Fig. 2). This group is paraphyletically positioned from the VN1 and the previously reported Asian haplogroup C [7].
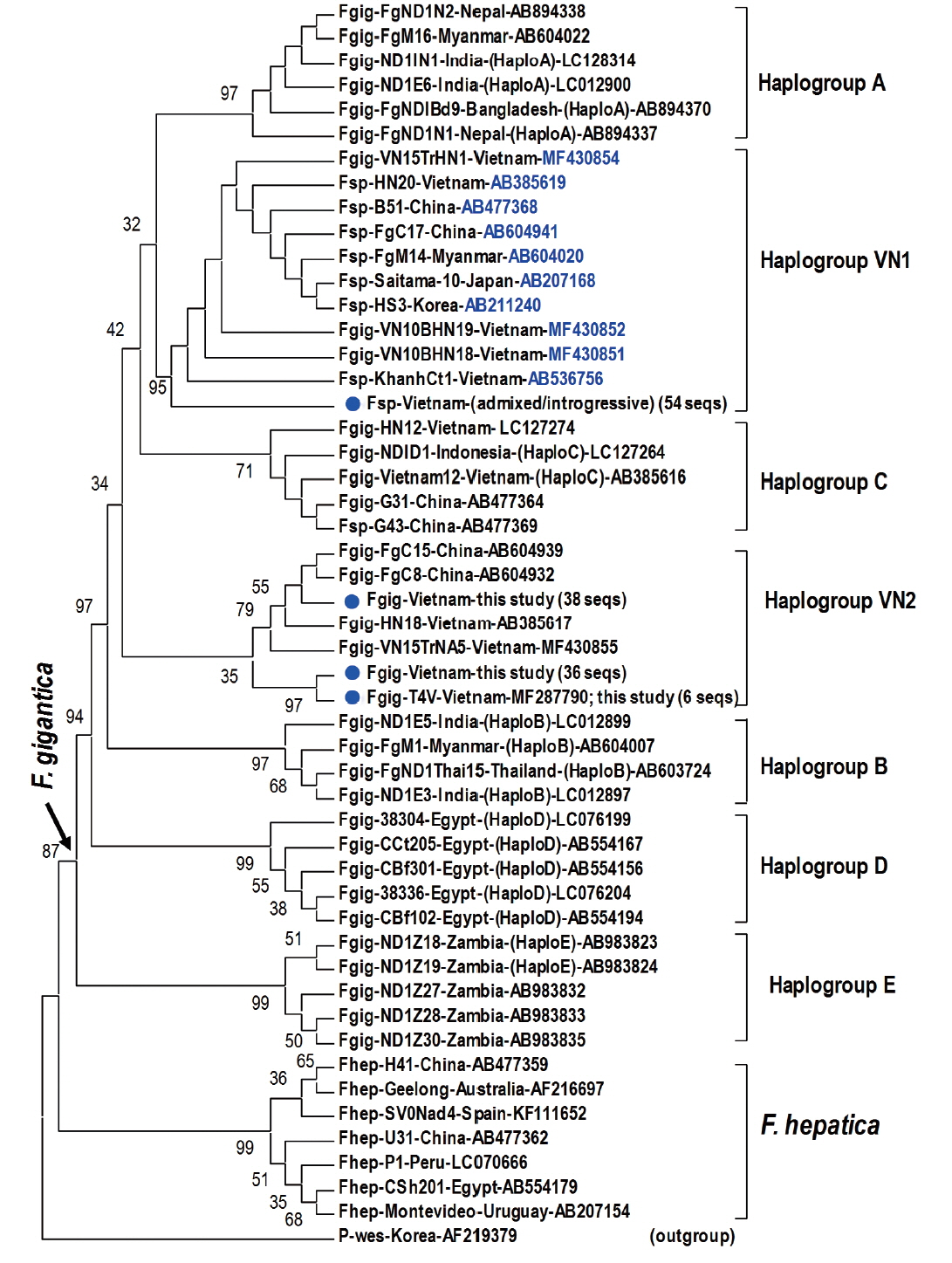
Phylogenetic tree showing topology of haplogroup relationships of Fasciola spp. from Vietnam and the reference isolates/strains from GenBank (Table 3) based on analysis of partial mitochondrial nad1 nucleotide sequence data (535 nucleotides) using Paragonimus westermani as an outgroup. Phylogenetic tree reconstruction was performed by MEGA 7.0 using a maximum likelihood (ML) analysis based on the general time-reversible model; supported for each node by 1,000 bootstrap resamplings [32]. In each sequence, species abbreviation is followed-up by strain and country (where the fluke was isolated). Names of haplogroups are included for reference (in brackets). Accession numbers are given at the end of each sequence. Basal node for F. gigantica is indicated by an arrow. Fasciola spp. sequences of this study from Vietnam are indicated by solid circles. Fsp: Fasciola species indicating intermediate forms (admixed/introgressive); number in bracket: identical nad1 sequences obtained in this study.
DISCUSSION
The present study was successful to examine morphological segregation of individual body shapes and to characterize molecular delineation of the interspecific hybridization based on species identification using nuclear and mitochondrial markers for Fasciola spp. in a mass of collected flukes from ruminants and samples from humans.
There have been numerous studies reporting the presence of, namely, intermediate forms of hybridization in the population of Fasciola spp. causing fascioliasis in ruminants and humans in 12 Asian and African countries [3–5,7–11,14,16,18,20,24]. The intermediate forms, genetically, are of the interspecific hybrid individuals between F. hepatica and F. gigantica which might result in the occurrence of aspermic parthenogenesis (asexual reproduction); and morphologically, they looked identical to F. hepatica flukes in the population [4,24]. None was any study in respect of the morphological appearance and molecular interrelationship for the prevalence of, and the indicative delineation for each, introgressive and admixed hybrid genotypes, among the intermediate Fasciola spp. in the fluke population.
The absence of the biologic taxon of F. hepatica was clearly defined. No sample having both, generic nuclear and mitochondrial genome, referring to F. hepatica, has been detected; although 284 F. hepatica-identical shape (6.0%) were examined in 4,725 flukes of ruminants in the present study [11,12,14,24]. However, single rDNA traits of F. hepatica were noted in 90% of the S-shaped and 15% of the M-shaped flukes (Table 5), giving the evidence for their introgression. We could not conduct the molecular analysis for all samples of 3, S-, M-, L-groups by using nuclear genes (protein-coding genes or their introns) described by [8,11]. However, the ITS and nad1 data in this study, as of being utilized in many previously reported implications, proved the reliable indication of the distribution of the hybrid types in Fasciola flukes in 18 Provinces in Vietnam. It also indicated that the use of the BL/BW ratio of body length and width should be cautious for discrimination between F. hepatica and F. gigantica [24,33], particularly, for F. hepatica-identical S-shape flukes, since their morphological values of F. hepatica (Fig. 1; Table 5) are contrasted to molecular data of their introgressive and admixed hybrid Fasciola spp. genotypes existed.
The co-existence of rDNAs from F. hepatica together with that of F. gigantica in 10% of S-shape and 7.5% of M-shaped flukes by molecular detection (Table 5) and this high percentage of the introgressive and admixed hybrid Fasciola spp. indicate the fact of considerable emergence of the intermediate Fasciola genotypes in ruminants in Vietnam. This rate was even higher, 22% (3/14 samples) for introgressive, and 14% (2/14) for admixed hybrid Fasciola spp. in the human specimens. However, the infective rate of introgressive/admixed hybrid Fasciola in humans does not mean their adaptation higher, since the specimens were only isolated on-site individually from the infected humans. Clarity of the hybrid status and the nationwide distribution of Fasciola spp. need a systematic study in combination of morphological and molecular approaches in Vietnam.
Topology of the nad1 phylogenetic tree in this study showed that the haplotypes of F. gigantica is diversified into 7 haplogroups. All the nad1 sequences of introgressive and admixed hybrid Fasciola come together constituting haplogroup VN1 and those of pure F. gigantica formed haplogroup VN2, both distinct from the previously reported haplogroup C. Apart from D (Egypt) and E (Zambia), the Asian haplogroups (A, B, C, and our additionally assigned VN1, VN2) seem to be a diverse complex. At the basal position in the tree, haplogroups C and VN2 indicated their F. gigantica provenance of the Asian origin. The haplotype VN1 of all hybridized Fasciola spp. from Vietnam, Korea, Japan, Myanmar and China, is separate from haplogroups B, C and VN2 (Fig. 2), suggesting that the introgressive and admixed hybrid Fasciola might originate from a distinct F. gigantica mtDNA lineage. F. gigantica of haplogroup C and VN2 played as an ancestral node for the divergence of the Asian F. gigantica-haplogroup A and the introgressive/admixed hybrid Fasciola-VN1 (Fig. 2).
Interspecies hybridization and introgression have become commonly detected in trematodes, first in Schistosoma spp. and Fasciola spp. due to intra and interspecific factors, referring to host-parasite and environmental changes [34,35]. Our findings of high prevalence of the introgressive/admixed hybrid Fasciola spp. in the absence of F. hepatica and their haplotypes from the distinct mtDNA lineage raise questions about the distribution of F. gigantica and both hybrid genotypes in other countries in/and out of Asia. The potential of hybrid Fasciola spp. to adapt in respect of infecting humans is also reasonably encountered. No link for the generic F. gigantica and the particular hybrid species determined yet due to the limited use of ITS1/2 and nad1 markers, thus, we need to obtain complete rDNA units and mtDNA genomes from 4 types, ie. F. hepatica, F. gigantica and each of introgressive/admixed hybrid Fasciola spp. for clear clarification.
ACKNOWLEDGMENTS
This work was supported by the National Foundation of Science and Technology of Vietnam (NAFOSTED) under grant number 106-YS.02-2013.06. We thank the laboratory technicians and veterinarians for their assistance in collection and preliminary physical grouping of the liver flukes; and hospital physicians for human specimens.
Notes
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interests related to this work.