Genetic Identification of Spirometra erinaceieuropaei Spargana in Liaoning and Hubei Provinces, PR China
Article information
Abstract
Spargana were collected from human and frogs in Liaoning and Hubei Provinces, China. PCR amplification and direct sequencing of A cox1 fragment was PCR-amplified from genomic DNA extracted from 7 specimens (5 from humans and 2 from frogs). The cox1 fragment (390 bp) showed 97–100% similarity to the reference sequence of S. erinaceieuropaei and 88–89% to the reference sequence of S. decipiens. There were 1–12 bases different between these worms, but no obvious genetic variation (0–3.3%) to the references. There was little difference of cox1 gene between sparganum samples of humans and frogs (1–3%). This study is the first report on S. erinaceieuropaei spargana from humans in Liaoning and Hubei Provinces.
Sparganosis is a zoonotic parasitic disease caused by the plerocercoid larvae (spargana) of the genus Spirometra, which belongs to the family of Diphyllobothriidae. Spargana can parasite within all vertebrate animals such as frogs, snakes, pigs, as well as human beings. The routes of infection involve either drinking natural water contaminated with procercoid-infected copepods or consumption of undercooked meat of plerocercoid-infected snakes or frogs. Additionally, infection can occur through skin wound by applying poultices such as skin or flesh of sparganum-infected snakes or frogs [1,2]. After infecting human, spargana migrate to multiple tissues and organs of the host and cause sparganosis. Dependent on the invaded tissue, spargana can cause blindness, limb paralysis, and even death [3–6].
Sparganosis has been reported worldwide, but most cases occur in several Asian countries such as China, Japan, South Korea and Thailand. Among all the countries, China has the largest number of sparganosis cases. Patients have been found in 27 provinces (mainly in coastal provinces such as Fujian, Guangdong and Zhejiang, as well as Sichuan and Hunan) since the first patient appeared in Xiamen in 1882. By 2014, there were more than 1,300 cases found national wide, composing more than 80% of global case reports [7].
At present, the species of the genus Spirometra, whose mitochondrial gene sequences have been determined, include S. erinaceieuropaei (S. erinacei), S. decipiens and S. ranarum. Reportedly, the first 2 kinds have been found and genetically identified in human patients in South Korea [8] and the last kind is found in Myanmar recently [9]. However, in China, the vast majority of human cases reported are confirmed as S. erinaceieuropaei based on morphological identification of the species. Identification of species at genetic level is primarily used on the animals. The prevalence of spargana infection in wild frogs (Rana rugulosa and R. temporaria) and snakes (Enhydris chinensis) was investigated in Guangxi Zhuang Autonomous Region of Southern China from June 2013 to August 2013. The results showed that 30.4% of the wild frogs and 50% of thesnakes were found infected with spargana and all the spargana were confirmed as of S. erinaceieuropaei [10,11]. Prevalence of spargana infection in the frogs (Rana nigromaculata) in Hunan, China was 20.2% [12]. A survey showed that sparganum infection rate was 51.9% in Rana tigrina rugulosa Wiegmann and 35.1% in Rana limnocharis Boie in Guangzhou [13]. Spargana of terrestrial snakes caught in Korea and China were identified as S. decipiens either by a multiplex PCR or mitochondrial cox1 sequence analyses [14].
So far, the analysis on spargana isolated from Liaoning and Hubei Provinces was limited to case reports or detecting infection rate. In addition, genetic analysis and exact identification evidence were inadequate. In other words, it was unclear whether they belong to S. erinaceieuropaei, S. decipiens or S. ranarum. Genetic identification on spargana of Spirometra species is, therefore, required.
In this study, we identified the 7 worms by observing the morphology and sequencing the cox1 gene amplified by NEST PCR. The spargana of human cases were collected from Xiaogan, Honghu and Xianning areas in Hubei, and Zhuanghe, Dandong areas in Liaoning, and wild frogs from Jinzhou National Park, Liaoning ICP: 09000177 (Table 1) from 2014 to 2018.

Information on samples used in this study
The worms were placed in a dish containing physiological saline, and general morphology of the worms was observed. Microstructures were observed on the specimens (H&E stained) under a microscope. All the worms were stored at 4°C in a 75% ethanol solution.
The worms were removed from the preservative and shredded by surgical scissors. Then, the genomic DNA was extracted by conventional phenol chloroform method after digested by SDS/ protein kinase K. At last, we used TE buffer solution to preserve the worms, at −20°C. The first set of PCR primers was p1f (5′-TGGTTTTTTGGACATCCTGAA-3′) and p1r (5′-ATCACATAATGAAAGTGAGCC-3′), which amplified a 440 bp product corresponding to the positions 707–1,146 bp of the cox1 gene. The second set of PCR primers, used for sequencing, was p1f1 (5′-GTGTTGATTTTGCCTGGGTTT-3′) and p1r1 (5′-TACAAACCAAGTATCATGTAA-3′), which yielded a 390 bp product corresponding to the positions 732–1,122 bp of the cox1 gene. These were designed from S. erinaceieuropaei (KJ599680) and S. decipiens (KJ599679). The PCR reaction was performed as follows: 1 cycle of initial denaturation at 94°C for 5 min, 35 cycles at 94°C for 1 min, 50°C for 1 min, 72°C for 1 min, and incubation at 72°C for 5 min. PCR were performed in a 50 μl reaction mixture with 4 μl of genomic DNA, 25 μl Premix dNTP, 2 μl of each primer (10 μmol), and 17 μl ddH2O. Double steamed water was used as blank control. Five microliter of the amplified products were identified as target genes by electrophoresis in 2% agarose gel. The remained 45 μl products were sent to Shanghai biotech Co., Ltd. for sequencing.
The Contig Express software (Informax Inc., Bethesda, Maryland, USA) was used to stitch the obtained sequences, and then the BioEdit software (Ibis Biosciences, Carlsbad, California, USA) was used for manual correction. The corrected sequences were identified by BLAST searches and compared with the homologous sequences published in GenBank. The cox1 gene sequences of other tapeworms belonging to the family Diphyllobothriidae was obtained from GenBank. The phylogenetic analysis was performed with MEGA version 7.0 by maximum likelihood (ML), maximum parsimony (MP), and neighbor-joining (NJ) methods, using Taenia solium as an outgroup. Clades were assessed by bootstrap resampling (1,000 replicates).
The spargana was 9.8–12.1 cm in length and 0.3–0.4 cm in width. All the samples had histological characteristics of sparganum: body folds, dense microtriches, calcareous corpuscles and muscle bundles.
The cox1 fragments (390 bp) amplified showed 97–100% similarity to the reference sequence of S. erinaceieuropaei (KJ599680.1) and 88–89% similarity to the reference sequence of S. decipiens (KJ599679.1). There were differences in the bases of the cox1 gene amplified between these worms (0–12), but no obvious genetic variation (0–3.3%). There was little difference of cox1 gene between human and frog (1–3%).
Phylogenetic trees constructed using MP, NJand ML methods were almost same, but bootstrap values were different. This paper mainly analyzed a phylogenetic tree of NJ method (Fig. 1). Liaoning isolates (LNZH1, LNDD1, LNJZ1 and LNJZ2) and Hubei isolates (HBXG1, HBHH1 and HBXN1) were clusted in a clade with an isolate of Korean cat (KJ599680.1) belonging to S. erinaceieuropaei. They were highly homologous and apart from branches of other species and genus tapeworms.
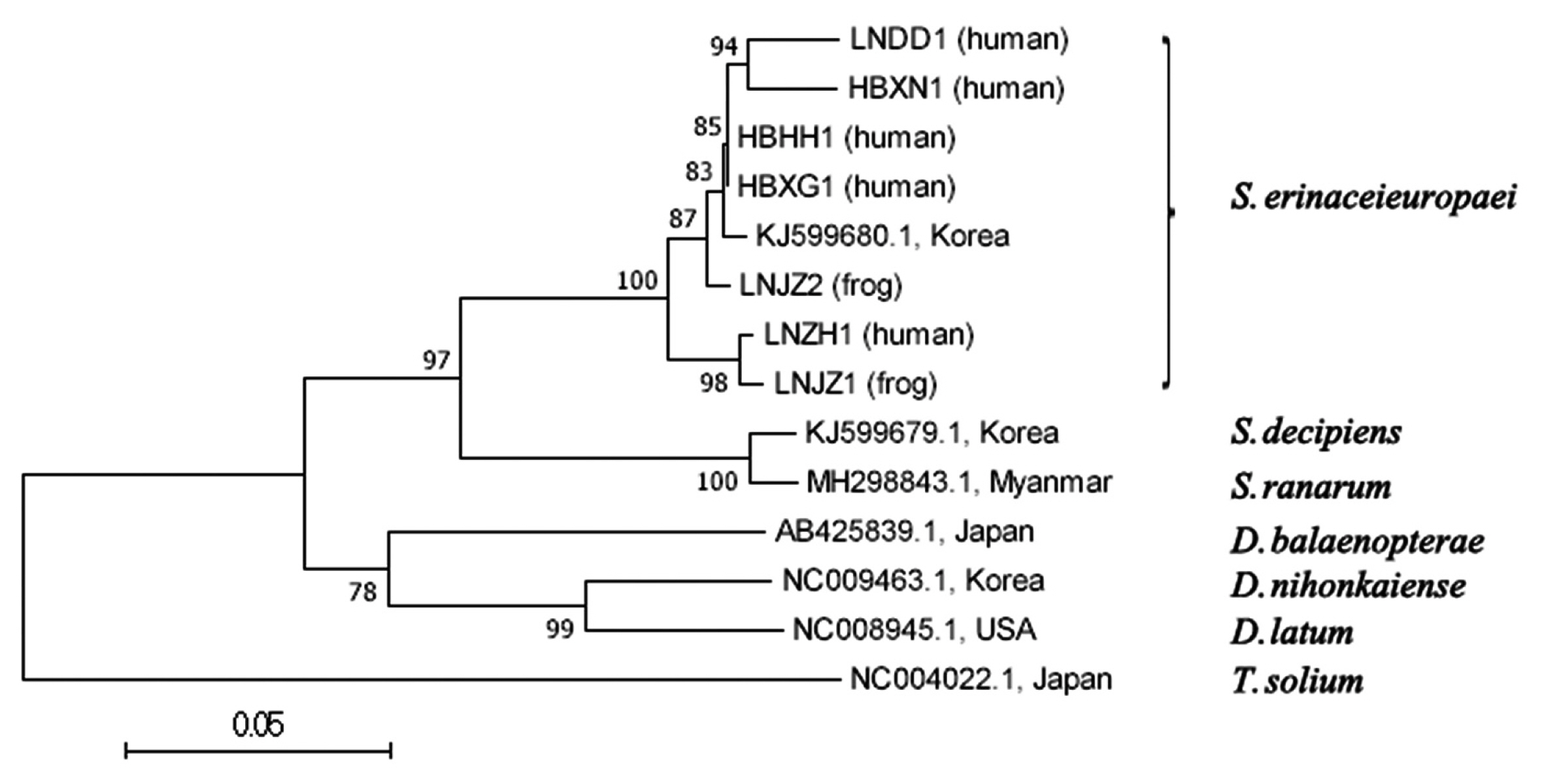
A phylogenetic tree using neighbor-joining method inferred from mitochondrial cox1 sequences of 7 Chinese cases and related Diphyllobothriidae species. Number at node indicates bootstrap value of 1,000 replicates.
The cox1 gene has been widely used in the research of genetic variation. For example, a case of a cat-derived spargana was confirmed as S. erinaceieuropaei in Iran [15]; a case of a dog-derived spargana was confirmed as S. decipiens in Australia [16]; all 904 samples of snake in China and South Korea were attributed to S. decipiens [14]. A domestic retrospective genetic analysis and sequencing analysis of 9 cases of human sparganosis in Hong Kong. The results showed that the 9 samples could be confirmed as S. erinaceieuropaei by the cox1 and 28S rRNA genes identification [17].
In this study, we found only one genotype in sequence variation analyses on the cox1 gene from 7 Spirometra specimens. The cox1 sequences (390 bp) of the 7 specimens showed high similarity to the reference sequence of S. erinaceieuropaei (KJ599680.1) and low similarity to the reference sequence of S. decipiens (KJ599679.1) and S. ranarum (MH298843.1). These results indicated that the examined Spirometra specimens in this study were identified as S. erinaceieuropaei by mitochondrial DNA sequence divergence. There was no obvious intraspecific variation (0–3.3%), but numerous differences between species (12–30%). The results of Okamoto (<2.6%) [18] and Liu (0–3.1%) [19] also indicated similar conclusion. Accordingly, we can conclude that the cox1 gene can provide genetic markers for genetic variation between species and can be used to identify Spirometra. The phylogenetic trees constructed by 3 different methods have consistency, and all of them showed that the isolates from both provinces were in the same large branch of S. erinaceieuropaei, which were far apart from the branches of other tapeworms.
Together with the identified taxonomic status, the findings of this paper can provide valuable information for further research on classification and epidemiology.
ACKNOWLEDGMENTS
We thank to Dr. Chun-Li An for invaluable advice and constant encouragement. We are grateful to Yao Fu for English proofreading. Approvals were given by the patients for the reporting of their case details. Funding source: This study was supported by grants from the National Natural Science Foundation of China (No. 81370189).
Notes
CONFLICT OF INTEREST
We have no conflict of interest related to this work.