Low Levels of Polymorphisms and Negative Selection in Plasmodum knowlesi Merozoite Surface Protein 8 in Malaysian Isolates
Article information
Abstract
Human infections due to the monkey malaria parasite Plasmodium knowlesi is increasingly being reported from most Southeast Asian countries specifically Malaysia. The parasite causes severe and fatal malaria thus there is a need for urgent measures for its control. In this study, the level of polymorphisms, haplotypes and natural selection of full-length pkmsp8 in 37 clinical samples from Malaysian Borneo along with 6 lab-adapted strains were investigated. Low levels of polymorphism were observed across the full-length gene, the double epidermal growth factor (EGF) domains were mostly conserved, and non-synonymous substitutions were absent. Evidence of strong negative selection pressure in the non-EGF regions were found indicating functional constrains acting at different domains. Phylogenetic haplotype network analysis identified shared haplotypes and indicated geographical clustering of samples originating from Peninsular Malaysia and Malaysian Borneo. This is the first study to genetically characterize the full-length msp8 gene from clinical isolates of P. knowlesi from Malaysia; however, further functional characterization would be useful for future rational vaccine design.
Plasmodium knowlesi, a zoonotic malaria parasite is now considered as the fifth Plasmodium species infecting humans as large number of cases have been reported from Southeast Asian countries, specifically Malaysia [1,2]. Within Malaysia, the highest number of human infections have been reported from Malaysian Borneo since 2004 [3–5] and very recently from Peninsular Malaysia too [6] highlighting the need of effective control measures and also the development of effective vaccines. With its 24-hr erythrocytic cycle, rapid increase in parasite counts has been shown to be associated with severe malaria and sometimes fatal [7,8]. Almost 70–78% of malaria cases reported from Malaysian Borneo (Sarawak and Kudat, Sabah) were due to P. knowlesi [5,9]. Recent genomic studies on P. knowlesi in clinical isolates of Malaysian Borneo have identified at least 3 sub-populations, which are highly diverse, 2 of the populations were associated with primary primate hosts and one with geographical location [10,11]. Analysis with mitochondrial genes in P. knowlesi clinical isolates and macaques also identified 2 distinct clusters which clustered geographically to Malaysian mainland and Malaysian Borneo [12].
Vaccine design and vaccine efficacy studies require the understanding of the extent and dynamics of genetic diversity in target antigens from malaria endemic regions. Major vaccine candidates studied in P. falciparum (like CSP, AMA1) show high genetic diversity and evolve under positive natural selection in the field in order to evade host immune pressure and thus are excellent targets for protective immunity but high variability also leads to non-efficacious vaccine trial due to strain-specific immune response [13]. For decades, vaccine research against malaria has primarily focused on P. falciparum and P. vivax and until date, not a single efficacious vaccine has been found which provides 100% protection. Merozoite surface proteins (MSPs) are recognized as potential vaccine candidates as they have been found to elicit a strong antibody response in patients and some molecules have shown strong inhibitory activity in RBCs in-vitro [14–16]. However, high antigenic variations within parasite populations are considered as one of the major hurdles in developing an efficacious and strain-transcending vaccine. Extensive genetic diversity has been observed in clinical isolates of P. knowlesi both in the genetic level as well as in the genomic level and several known ortholog vaccine antigens have shown similar levels of high diversity [10,11,17–20]. These studies highlight the complexities involved in P. knowlesi vaccine design thus a rational approach would be necessary. Thus, it is important to identify potential blood stage parasite antigens, which are essential for its survival, low in polymorphism in the endemic region and show significant immune response in patient serum. Merozoite surface protein 8 (MSP8) contains 2 copies of a conserved epidermal growth factor (EGF)-like domain at the carboxyl terminal that is anchored to the membrane via glycosylphosphatidylinositol (GPI) membrane anchor [17]. Specific binding of MSP8-peptides to human RBCs have been reported in P. falciparum suggesting an essential role in parasite invasion [21] and naturally acquired humoral and cell mediated immune response in patient sera have been observed in P. vivax [22]. Genetic studies on MSP8 of P. falciparum and P. vivax from world-wide isolates (from different geographical locations) indicated that the gene is under purifying selection and has low levels of polymorphism [23]. However, no studies have been conducted in Pkmsp8 from clinical samples. Thus, this study was designed to determine the level of diversity, haplotypes and natural selection acting at the full-length gene and its domains from clinical isolates from Sarawak, Malaysian Borneo.
Pkmsp8 sequences were downloaded for 37 clinical isolates originating from Kapit, Betong and Sarikei from Sarawak, Malaysian Borneo along with 6 long-time isolated lines originated from Mainland Malaysia along with the H-strain (PKNH_ 1031500) [11]. The sequence data with accession numbers are same as used for a previous study [17]. The PkMSP8 domains were characterized based on the published ortholog in PvMSP8 (PVX_097625) [22]. Sequence diversity (π), the number of polymorphic sites, number of synonymous and non-synonymous substitutions, haplotype diversity (Hd) and number of haplotypes (H) within the pkmsp8 sequences was determined by DnaSP v5.10 software [24]. Natural selection was determined at the intra-population level by calculating the rates of synonymous substitutions per synonymous site (dS) and non-synonymous substitutions per non-synonymous site (dN) were computed by using Nei and Gojobori’s method and robustness were estimated by the bootstrap method with 1000 pseudo-replicates as implemented in the MEGA 5.0 software [25]. If dN-dS differences were positive, it corresponds to positive natural selection and negative values corresponds to negative selection. Also, codon based Z-test for selection was implemented with MEGA software to test the significance using 1000 bootstrap values. Tajima’s D, Fu & Li’s D* and F* tests were implemented in DnaSP v5.10 software. When Tajima’s D, Fu & Li’s D* and F* values are positive and significant, it indicates positive/balancing selection, whereas negative values suggest negative selection or population expansion. To test whether the pkmsp8 gene is under the influence of natural selection in the inter-species level, the robust McDonald and Kreitman (MK) test were performed with both P. coatneyi (PCOAH_00031550) and P. cynomolgi (PCYB_104050) msp8 gene as an out-groups using DnaSP v5.10 software [25]. Genealogical relationships were constructed between the pkmsp8 haplotypes using the median-joining method in NETWORK software (version 4.6.1.2, Fluxus Technology Ltd., Suffolk, UK).
The schematic structure of pkmsp8 gene and its domains were demarcated based on the P. vivax MSP8 protein (Fig. 1A). There was an asparagine-rich region (ASN) upstream the double EGF domains, which was similar to P. vivax MSP8 [22] (Fig. 1A). Within the full-length pkmsp8 sequences (n=43, 1,431 bp), there were 24 polymorphic sites (1.67%) leading 17 synonymous and 7 non-synonymous substitutions. We found 8 parsimony informative sites and 16 singleton variable sites. The overall nucleotide diversity was lower (π=0.0018±SD 0.00028) compared to its ortholog in P. vivax and P. falciparum which is relatively conserved (Table 1) [23]. Low levels of polymorphism were observed across the full-length pkmsp8 gene and the majority of the SNPs were synonymous substitutions. Similar reports of high number of synonymous substitutions in msp1p and msp1 genes were observed in P. knowlesi clinical isolates with negative/purifying natural selection [20,26]. The diversity towards the non-EGF and the double EGF domains were of similar levels (π=0.00010–0.00016), however, the number of SNPs towards the non-EGF domains was higher than EGF domain (Table 1) indicating the EGF domains were conserved. Sequence alignment showed that non-synonymous substitutions were absent within the C-terminal double EGF domains compared with the non-EGF domain (Table 1). It is to be noted that the singleton sites were scattered more towards the non-EGF domain (Si=13), while the EGF domains had only three. The sliding window plot analysis (window length 100 bp and step size 25 bp) also revealed that the overall diversity ranged from 0 to 0.0061 and the C-terminal double EGF domains containing the 19 kDa domain showed lower diversity (Fig. 1B). Due to the high number of singletons in the non-EGF domains, the number of haplotypes and haplotype diversities were higher compared to the EGF domain within the pkmsp8 gene (Table 1). The 12-cysteine residues within the 2 EGF domains at the 19 kDa domain were conserved within the clinical isolates indicating functional conservation.
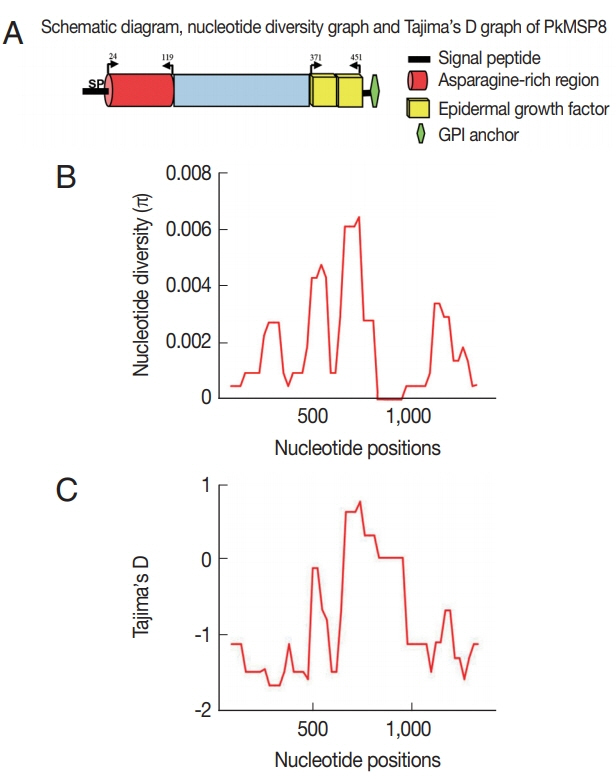
(A) Schematic diagram of Plasmodium knowlesi MSP8 domains (B) Graphical representation of nucleotide diversity (π) within 37 full-length pkmsp8 genes (n=1,431 bp) from Malaysian Borneo and the lab-adapted strains. The PkMSP8 domains are marked above. (C) Graphical representation of Tajima’s D value across the MSP8 gene.

Estimates of nucleotide diversity, natural selection, haplotype diversity and neutrality indices of pkmsp8 and its domains within 43 isolates
To determine whether natural selection contributes to the polymorphism in the pkmsp8 full-length gene as well as at each domain (EGF and non-EGF), multiple tests were conducted; at the intra-species level significant negative value was observed at the full-length gene and at the non-EGF domain (Table 1) indicating dN<dS. Additional statistical test for neutrality; Tajimas’D, Li and Fu’s F* and D* statistics also showed significant negative values for the full-length gene and the non-EGF domain indicating negative/purifying natural selection and parasite population expansion in Malaysian Borneo. However, it was observed that even though test results were with negative values for the double EGF domain, it was not statistically significant and the absence of non-synonymous substitutions within the double EGF domain indicate an absence of natural selection within the domain and functional conservation. Interestingly, all the 12 cysteine residues within the double EGF domains were conserved within the 43 isolates (including the lab adapted strains) indicating conserved functional activity. This conservation of the 12 cysteine residues was also observed for the C-terminal 19 kDa fragment of PvMSP1P and PkMSP1P [26] and binding activity to reticulocytes in P. vivax have been reported [14,27–29]. Sliding window plot analysis of Tajima’s D across the full-length pkmsp8 gene also indicated most values below 0 indicating purifying selection (Fig. 1C). Natural selection test using the MK test (at the inter-species level) with P. coatneyi and P. cynomolgi as orthologous sequences also showed similar results with strong negative/purifying selection acting at the full-length and the non-EGF domain probably due to functional constraints and the domain may not be exposed to host immune pressure (Table 2).

McDonald–Kreitman tests on MSP8 of Plasmodium knowlesi and its domains with P. coatneyi and P. cynomolgi orthologs as outgroup sequences
Two distinct population clusters were observed; one originating from Malaysian mainland where the lab adapted strains originated (i.e., H, Malayan, Nuri, Hackeri, Philippine and MR4H) and the other sub-cluster was the clinical isolates from Sarawak, Malaysian Borneo (Fig. 2). Two major shared haplotypes were found between parasite populations from Kapit, Betong and Sarikei (H_5 and H_6) (Fig. 2) and these 2 haplotype clusters may represent the two distinct clusters observed from Malaysian Borneo. Similar findings with clinical isolates from Malaysian Borneo have been reported earlier with merozoite surface proteins [17,20,26,30]. It is interesting to note that MSP8 gene has very low levels of polymorphism compared to its orthologs in P. vivax and P. falciparum [23] and thus might be an ideal candidate for vaccine design against P. knowlesi. This may be due to the absence of host immune pressure in the EGF domains which is critical for binding to host cells. However, further studies characterizing the immunological and functional validation of the candidate would be necessary.
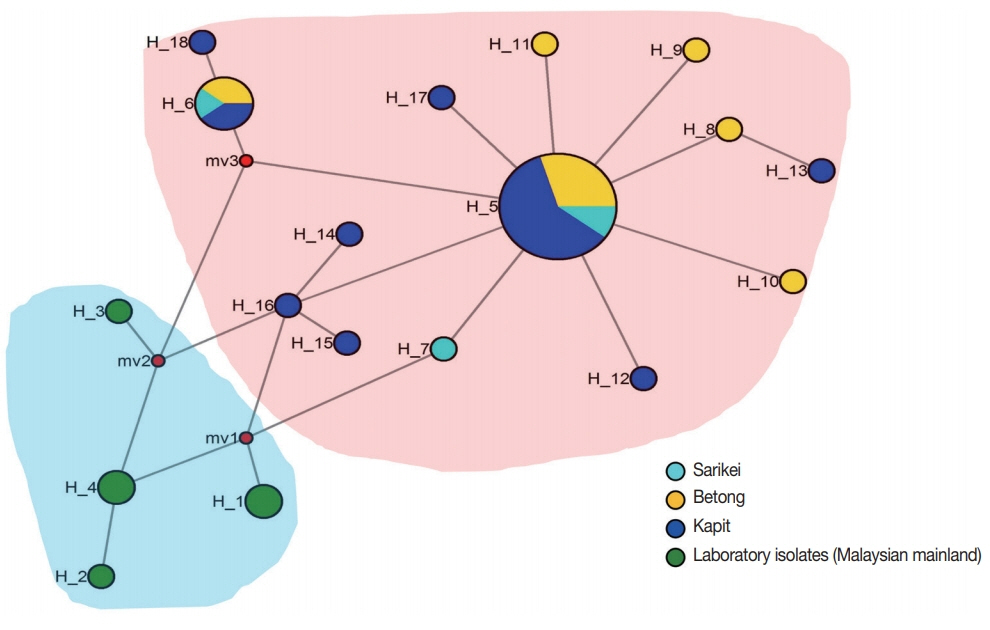
Median-joining networks of Plasmodium knowlesi MSP8 haplotypes from Malaysia. The genealogical haplotype network shows the relationships among the 18 haplotypes present in the 43 sequences obtained from clinical isolates and lab adapted strains from 5 geographical regions of Malaysia. Each distinct haplotype has been designated a number (H_n). Circle sizes represent the frequencies of the corresponding haplotype (the number is indicated for those that were observed >1×). Distances between nodes are arbitrary. The small red circles are median vectors randomly generated by the software while constructing the network.
ACKNOWLEDGMENT
This work was supported by grants from the National Research Foundation of Korea (NRF) (2018R1A2B6003535, 2018R1A6A1A03025124).
Notes
CONFLICT OF INTEREST
The authors declare that they have no competing interests.