Abstract
Toxoplasma gondii, an intracellular protozoan parasite that infects one-third of the world’s population, has been reported to hijack host cell apoptotic machinery and promote either an anti- or proapoptotic program depending on the parasite virulence and load and the host cell type. However, little is known about the regulation of human FHs 74 small intestinal epithelial cell viability in response to T. gondii infection. Here we show that T. gondii RH strain tachyzoite infection or ESP treatment of FHs 74 Int cells induced apoptosis, mitochondrial dysfunction and ER stress in host cells. Pretreatment with 4-PBA inhibited the expression or activation of key molecules involved in ER stress. In addition, both T. gondii and ESP challenge-induced mitochondrial dysfunction and cell death were dramatically suppressed in 4-PBA pretreated cells. Our study indicates that T. gondii infection induced ER stress in FHs 74 Int cells, which induced mitochondrial dysfunction followed by apoptosis. This may constitute a potential molecular mechanism responsible for the foodborne parasitic disease caused by T. gondii.
-
Key words: Toxoplasma gondii, ER stress, mitochondrial apoptosis, FHs 74 Int cell
INTRODUCTION
Toxoplasma gondii (
T. gondii) is one of the world’s most successful parasites, in part because of its ability to infect and persist in most warm-blooded animals [
1]. Natural infection by
T. gondii occurs via oral ingestion of cyst-containing meat or oocyst-contaminated water and produce. As a result, the first encounter between parasite and host occurs in the small intestinal epithelium [
2]. Tissue cysts that rupture in the small intestine and release zoites infect locally before disseminating throughout the host, where they form cysts that persist for the lifetime of the infected individual. This can have serious consequences to human health: reactivation of cysts in people whose immune systems are compromised can result in severe encephalitis and death [
3]. Furthermore, spontaneous abortion, stillbirth, and severe birth defects can occur if the infection occurs during pregnancy and is transmitted to the fetus [
4].
Excretory/secretory proteins (ESP) from
T. gondii plays an important role in generating suitable conditions for parasite invasion into host cells [
5]. One of the mechanisms by which the parasite escapes host immune responses is antigen shedding, a mechanism in which parasite ESP is released into the host body and evokes the immune system [
6]. Apoptosis is a form of programmed and controlled cell death that accounts for the majority of cellular death in bioprocesses [
7]. Mitochondria is important in the regulation and transmission of apoptotic signals, which are regulated by maintaining a balance between the levels of Bcl-2-family proteins [
8].
T. gondii can modulate the host cell response, and apoptosis is one of the mechanisms targeted by this parasite [
9]. A recent study revealed that
T. gondii triggers trophoblast apoptosis through oxidative stress and mitochondrial dysfunction in mice [
10]. Infection of murine astrocytes by the highly virulent RH strain led to decreased expression of the antiapoptotic proteins survivin, p53 upregulated modulator of apoptosis (PUMA), and Bcl-2 and increased expression of the proapoptotic protein Noxa in the early stage of infection [
11]. Conversely,
T. gondii and ESP have the ability to inhibit apoptosis in several murine and human host cells treated with a broad spectrum of proapoptotic stimuli [
12–
14]. Presumably,
T. gondii interferes with different processes to inhibit apoptosis and maintain chronic infection.
Bcl-2 plays an important role in the maintenance of mitochondrial integrity by preventing apoptosis-induced cytochrome c release, and cytochrome c is involved in activating downstream caspases that trigger apoptosis [
15]. Several in vitro studies have reported that
T. gondii infection regulates mitochondrial apoptosis through modulation of Bcl-2 family protein expression [
10,
13,
14]. The endoplasmic reticulum (ER) functions in protein folding and assembly, lipid biosynthesis, vesicular trafficking, and cellular calcium storage. Its function can be disturbed by various factors, such as inhibition of protein glycosylation, calcium depletion, changes in redox status, and the expression of misfolded proteins. These dysfunctions cause proteotoxicity in the ER, collectively termed ER stress, which leads to activation of the unfolded protein response (UPR) [
16,
17].
T. gondii induces apoptosis of neural stem cells via the endoplasmic reticulum stress pathway [
18]. However, little is known about the molecular mechanism of apoptosis in human small intestinal epithelial cells induced by
T. gondii infection.
Here, we aimed to investigate the possible implication of endoplasmic reticulum stress-mediated mitochondrial apoptosis and its mechanisms in T. gondii-infected and ESP-treated FHs 74 Int cells. FHs 74 Int cells were infected with T. gondii or treated with ESP alone or in combination with the ER stress inhibitor 4-phenylbutyric acid (4-PBA), and then, cellular cytotoxicity, apoptosis, Bcl-2 family members and ER stress signaling pathways were evaluated.
MATERIALS AND METHODS
Reagents and antibodies
A CytoTox 96® Non-Radioactive Cytotoxicity Assay was obtained from Promega (Madison, Wisconsin, USA). A JC-1 MitoMP detection kit was obtained from Dojindo (Kumamoto, Kumamoto, Japan). The ER stress inhibitor 4-phenylbutyrate (4-PBA) was purchased from Sigma Chemical Co. (St. Louis, Missouri, USA).
An ER Stress Antibody Sampler Kit; anti-activating transcription factor 6 (ATF6), anti-phospho protein kinase RNA-like endoplasmic reticulum kinase (p-PERK), anti-phospho eukaryotic translation initiation factor 2α (p-eIF2α), anti-poly (ADP-ribose) polymerase (PARP), and anti-cleaved caspase-3 antibodies; and a pro-Apoptosis Bcl2 Family Member Antibody Sampler Kit and pro-Survival Bcl-2 Family Member Antibody Sampler Kit were purchased from Cell Signaling Technology Inc. (Danvers, Massachusetts, USA). Anti-CCAAT/enhancer-binding protein–homologous protein (CHOP) and anti-phospho inositol-requiring protein 1α (p-IRE1α) antibodies were obtained from Abcam (Cambridge, Massachusetts, USA). Secondary anti-rabbit-horseradish peroxidase (HRP) and anti-mouse-HRP antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, Pennsylvania, USA).
Culture of FHs74 Int cells
A nontransformed human fetal small intestinal epithelial cell-line (FHs 74 Int cells) was purchased from ATCC (ATCC, Manassas, Virginia, USA) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), antibiotic-antimycotic (Gibco BRL) and 30 ng/ml human epidermal growth factor (all from Gibco, Grand Island, New York, USA) at 37°C in a humidified atmosphere with 5% CO2. The medium was changed every 2–3 days.
Maintenance of T. gondii
Tachyzoites of the
T. gondii RH strain was maintained as described previously [
19]. Briefly, human retinal pigment epithelial (ARPE-19) cells (ATCC) were cultured in a 1:1 (v/v) mixture of DMEM/F12 supplemented with 10% heat-inactivated FBS and an antibiotic-antimycotic solution. ARPE-19 cells were infected with
T. gondii at a multiplicity of infection (MOI) of 5 for 2–3 days. After spontaneous host cell rupture, parasites and cellular debris were pelleted by centrifugation and washed in cold PBS. The final pellet was resuspended and passed through a 26-gauge needle fitted with a filter with 5.0-μm pores (Millipore, Billerica, Massachusetts, USA).
Toxoplasma gondii ESP was produced as described previously [
20] with slight modifications. Briefly, freshly purified tachyzoites (1×10
8) from the peritoneal cavity were incubated at 37°C for 3 hr under mild agitation in test tubes containing 1.0 ml Hank’s balanced salt solution (HBSS) (Gibco BRL, Rockville, Maryland, USA). After centrifugation for 5 min at 6,000 g, the supernatant, including ESP, was saved. The protein concentration was measured using a Bradford assay (Bio-Rad Laboratories, Hercules, California, USA), and samples were stored at −70°C until use.
Experimental designs
FHs74 Int cells were seeded on 96-well plates (for LDH and MTS assays), 12-well coverslips (for immunofluorescence), and 100-mm culture dishes (for western blotting) at various densities and grown to confluence at 37°C in 5% CO2.
FHs74 Int cells were stimulated with tachyzoites of the T. gondii RH strain at MOIs of 10 or with 1 μg/ml ESP for 0, 2, 8, and 24 hr. Then, cell cytotoxicity, apoptosis, apoptotic features, induction of ER stress and its mechanism of action, and Bcl-2 family-related protein expression were evaluated using a LDH assay, immunocytochemistry and western blotting.
To determine the role of ER stress in T. gondii- or ESP-challenged cells, FHs74 Int cells were pretreated with the ER stress inhibitor 4-PBA and then evaluated for an ER stress response, apoptotic features, MMP and Bcl-2 family protein expression. Untreated FHs74 Int cells were used as controls. Each experiment was performed at least 3 times in triplicate.
LDH Assay
The LDH assays were performed for cytotoxicity quantification using a CytoTox 96® Non-Radioactive Cytotoxicity Assay kit according to the manufacturer’s protocol. Briefly, 1×104 FHs74 Int cells were seeded in 96-well plates and infected with T. gondii at an MOI of 10 or treated with 1 μg/ml ESP for the indicated times (0, 2, 8, and 24 hr) in an incubator (5% CO2, 90% relative humidity, 37°C). Then, 50 μl aliquots from all infected and control wells were transferred into a new 96-well plate, and 50 μl of CytoTox 96® reagent was added to each sample aliquot. The plate was covered with foil to protect the samples from light and incubated for 30 min at room temperature. After addition of 50 μl of stop solution to each well, the absorbance of the solution was measured immediately at 490 nm using a microplate reader (TECAN, Crailsheim, Germany). LDH release levels in the medium were quantified and compared to control values according to the kit instructions.
MMP Assay
MMP was measured using a JC-1 MitoMP detection kit (Dojindo, Kumamoto, Japan). Briefly, FHs74 Int cells were seeded onto coverslips in 12-well plates at a density of 1×104 cells/well and infected with T. gondii at an MOI of 10 or treated with 1 μg/ml ESP under various conditions with or without an ER stress inhibitor. The cells were then incubated with 4 μm JC-1 fluorescent dye at 37°C for 30 min in the dark and rinsed 3 times with HBSS. The stained cells were mounted onto microscope slides in VECTASHIELD HardSet Mounting Medium with DAPI (Vector Laboratories), and images were collected using a laser confocal microscope (Leica, TCS SP8, Wetzlar, Germany). The intensities of green (excitation/emission wavelength=485/538 nm) and red (excitation/emission wavelength=485/590 nm) fluorescence were analyzed in ≥6 microscopic fields in each sample. The intensities of red and green fluorescence were calculated using Image J software (NIH, Bethesda, Maryland, USA).
Western blotting analysis
SDS-PAGE and western blotting analysis were performed to determine the expression of numerous proteins. FHs74 Int cells were cultured in 100-mm dishes and underwent serum deprivation for 4 hr to remove stimulation from serum factors. Then, the cells were stimulated with T. gondii or ESP as indicated. After washing with PBS, proteins were extracted using PRO-PREP Protein Extraction Solution (iNtRON Biotechnology, Korea). The extract was incubated with complete protease inhibitor cocktail (Roche, Basel, Switzerland) for 30 min on ice followed by boiling for 10 min and then centrifuged at 14,000 g for 15 min at 4°C. The supernatant was collected, and equal amounts of protein from each sample were separated via SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad). For blocking, the membranes were immersed for 1 hr at room temperature in 5% skim milk in Tris-buffered saline (20 mM Tris, 137 mM NaCl, pH 7.6) containing 0.1% Tween-20 (TBST). After one wash in TBST, the membranes were incubated overnight at 4°C with primary antibodies diluted in TBST supplemented with 5% BSA. Following 3 consecutive washes in TBST, membranes were incubated for 90 min with HRP-conjugated anti-mouse or anti-rabbit secondary antibody diluted 1:5,000 with 5% skim milk, as described above. The membrane was soaked with Immobilon Western Chemiluminescent HRP Substrate (Jackson ImmunoResearch Laboratories), and chemiluminescence was detected using a Fusion Solo System (Vilber Lourmat, Collegien, France). These experiments were repeated at least 3 times. The band intensities were quantified using ImageJ software (NIH). The results were normalized to α-Tubulin protein levels and were expressed as fold changes over the mock-infection control group.
Preparation of mitochondrial and cytosolic fractions
Mitochondrial and cytosolic fractions of FHs 74 Int cells were prepared as described previously [PMID: 32547023]. For cytosolic extracts free of nuclei and mitochondria, the cells were washed in ice-cold PBS (pH 7.2) and then in hypotonic extraction buffer (HEB; 50 mM PIPES, 50 mM KCl, 5 mM EGTA, 2 mM MgCl2, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, pH 7.4) and harvested by centrifugation. Pellets were resuspended in HEB and lysed with a Dounce homogenizer. These cell lysates were then centrifuged at 100,000×g for 60 min at 4°C, and the supernatants were flash-frozen in cold ethanol, aliquoted, and stored at −80°C. Mitochondrial fractions were prepared by washing the cells in ice-cold PBS and then resuspending them in an isotonic homogenization buffer (10 mM Tris-HCl, pH 7.5, 250 mM sucrose, 10 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, EDTA-free complete cocktail of protease inhibitors [Roche]). After 60 strokes in a Dounce homogenizer, unbroken cells were removed by centrifugation at 1,000×g for 10 min, and the supernatants were centrifuged at 14,000×g for 20 min. The supernatants were collected as the mitochondrial fraction. The expression of cytochrome c expression was determined by western blotting. COX IV and α-tubulin were used as mitochondria and cytosol markers, respectively.
Statistical analysis
All assays were performed in triplicate, and at least 3 independent experiments were conducted per test series. The results are presented as mean±SD. Statical analysis of the data was performed using unpaired, 2-tailed Student’s t-tests. A P-value less than 0.05 indicated statistical significance.
RESULTS
T. gondii infection and ESP treatment induced cytotoxicity and apoptosis to FHs 74 Int cells
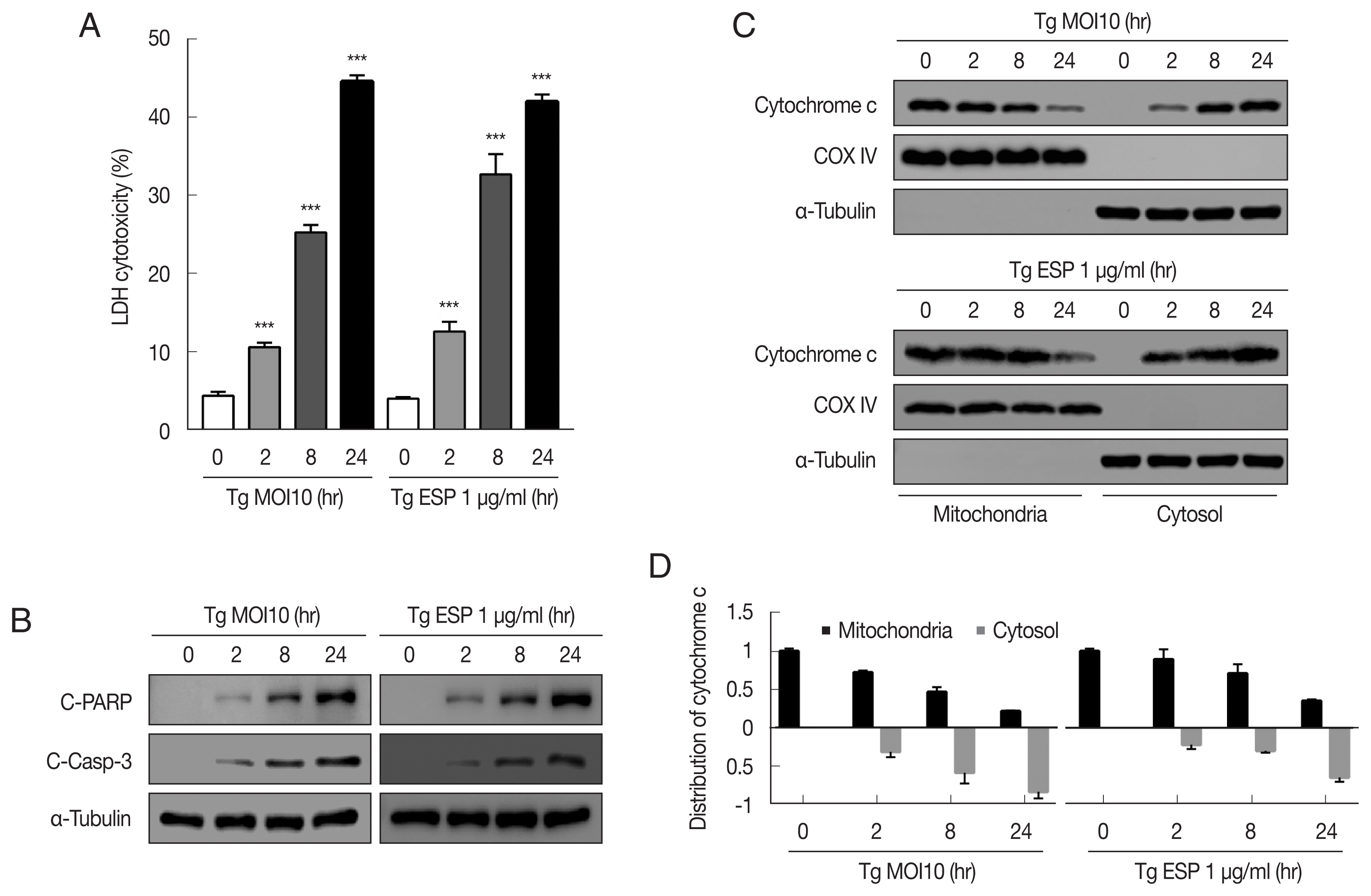
The cytotoxicity of
T. gondii RH tachyzoites to FHs 74 Int cells were 4.25±0.63%, 10.33±0.46%, 24.72±0.31%, and 44.42±1.05% at 0, 2, 8, and 24 hr postinfection, respectively. Next, we examined whether
T. gondii ESP is toxic to FHs 74 Int cells. ESP (1 μg/ml) was added to the host cells for the indicated time, and the cytotoxicity was dramatically increased to 12.62±1.25%, 32.68±2.57%, and 41.98±0.94% at 2, 8, and 24 hr, respectively (
Fig. 1A). To verify whether the cytotoxicity of the
T. gondii RH strain or ESP to FHs 74 Int cells was associated with apoptosis, we measured the level of apoptosis indicator proteins in RH-infected or ESP-treated FHs 74 Int cells. Our results showed that the protein levels of cleaved PARP and cleaved caspase 3 were significantly elevated in response to RH and ESP, and their increase was time dependent (
Fig. 1B). Similarly,
T. gondii infection and ESP treatment time-dependently elevated release of cytochrome c from mitochondria into cytoplasm (
Fig. 1C, D). Collectively, our data indicate that
T. gondii RH strain tachyzoites and ESP induced cytotoxicity and apoptosis in FHs 74 Int cells in a time-dependent manner.
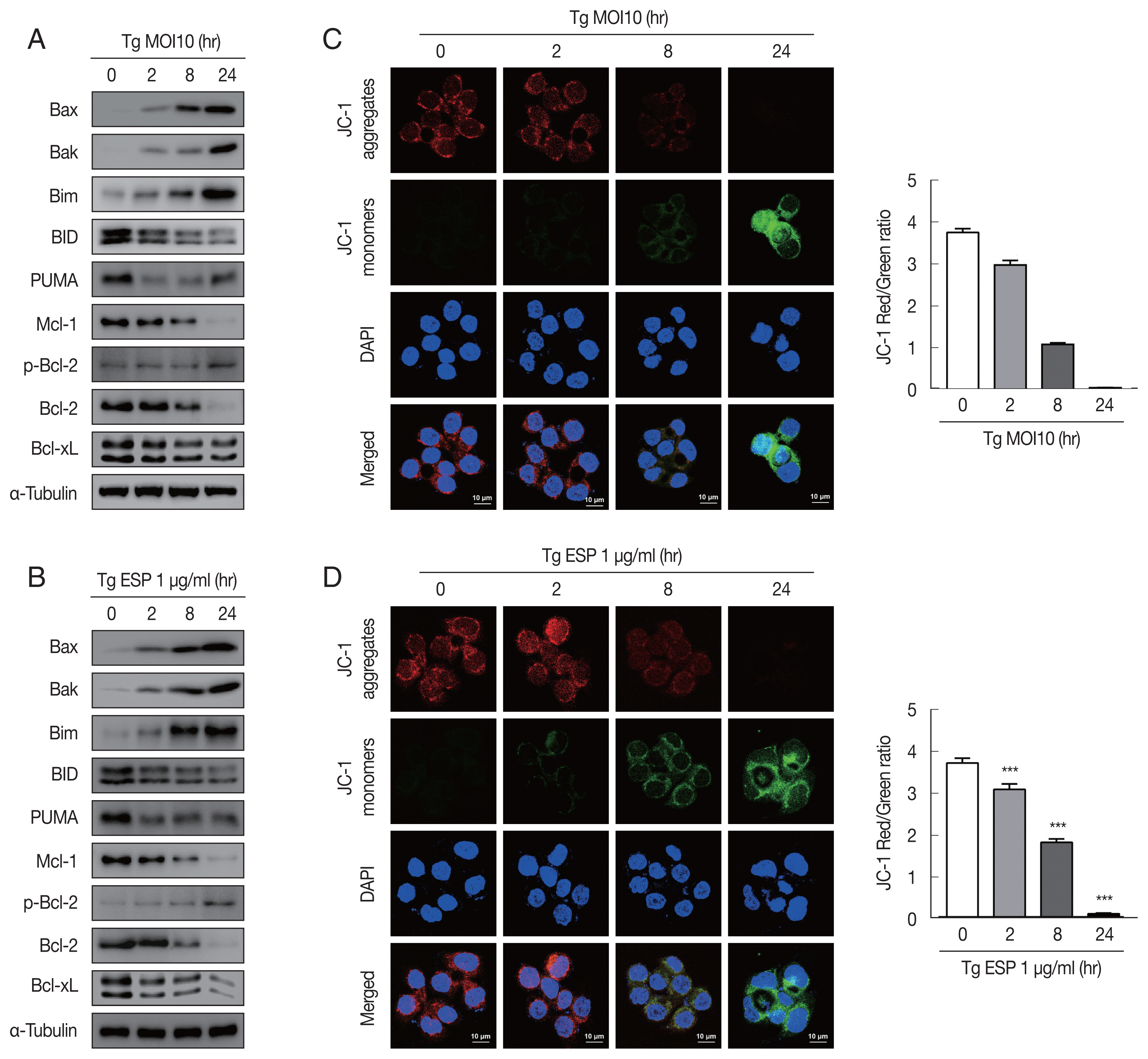
Western blot analysis data showed that the protein levels of Bax, Bak and Bim were obviously increased with
T. gondii infection, whereas the protein levels of BID, Mcl-1, Bcl-2, and Bcl-xL were drastically reduced in a parasite infection time-dependent manner (
Fig. 2A). JC-1 dye staining revealed that
T. gondii time-dependently induced MMP depolarization (
Fig. 2C).
Upon ESP treatment, the protein levels of Bax, Bak, and Bim were apparently increased and those of BID, Mcl-1, Bcl-2, and Bcl-xL were diminished in a time-dependent manner (
Fig. 2B). Surprisingly, we also found that the PUMA protein level was reduced after 2 and 8 hr of
T. gondii infection and then slightly increased at 24 hr (
Fig. 2A), but with ESP treatment, PUMA was dramatically reduced at 2 hr and remained low until 24 hr (
Fig. 2B). In addition, both
T. gondii and ESP induced modest phosphorylation of Bcl-2 at 24 hr (
Fig. 2A, B). ESP treatment also caused mitochondrial dysfunction in a time-dependent manner (
Fig. 2D). These findings suggest that
T. gondii RH strain tachyzoites derived mitochondrial dysfunction in FHs 74 Int cells in a time-dependent manner. Speculation is not Results.
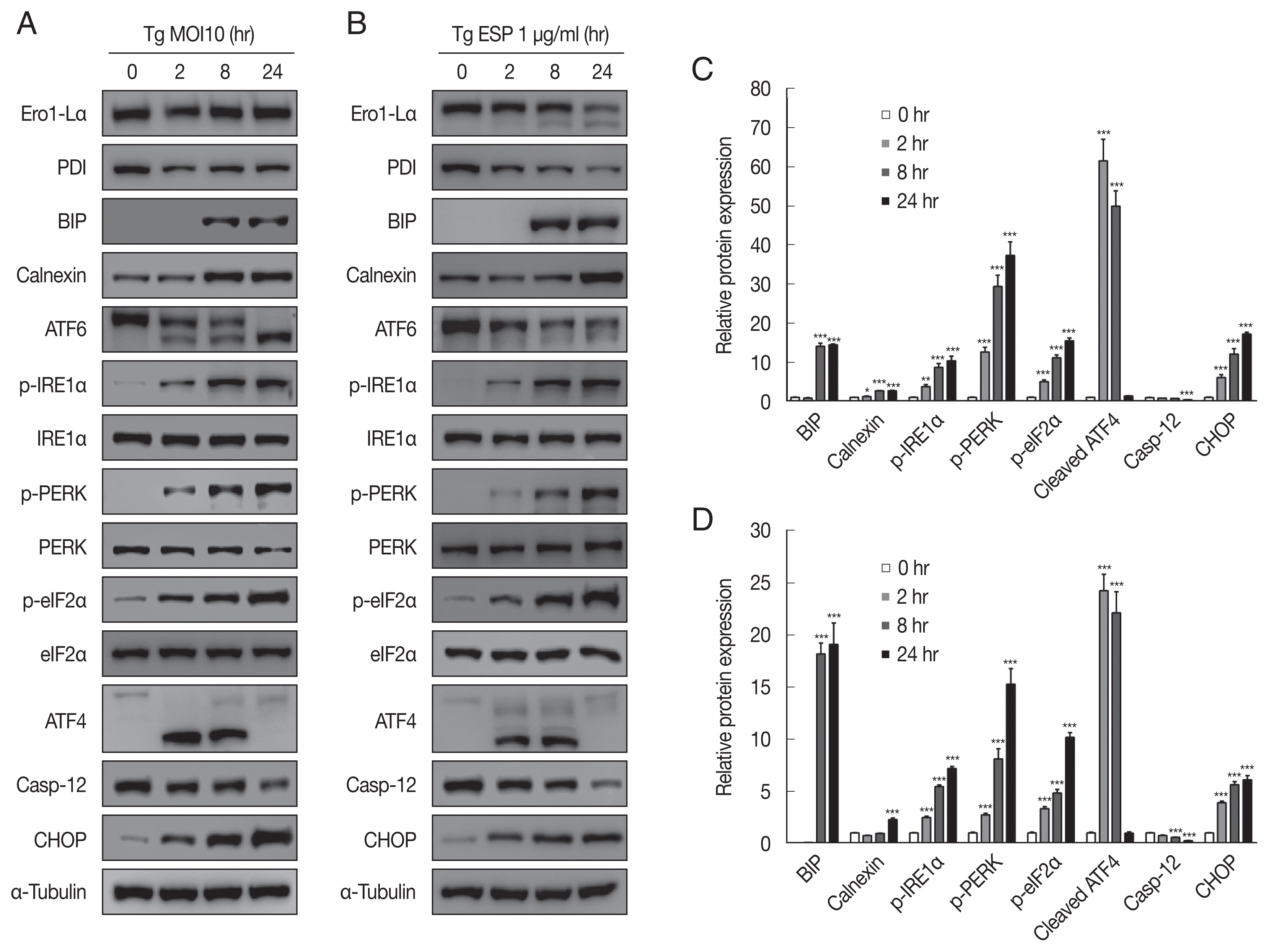
The ER-resident transmembrane sensor proteins IRE1, PERK and ATF6 were activated, evidenced by the dramatic increase in phospho-IRE1α, phospho-PERK, phospho-eIF2α and cleaved ATF6 upon
T. gondii infection (
Fig. 3A, C). CHOP, which plays an important role in ER stress-induced apoptosis, was also elevated. However, the protein level of caspase 12, which is essential for carrying out the caspase-dependent UPR, was visibly reduced compared with that in the uninfected control group. All the above events occurred in a parasite infection time-dependent manner (
Fig. 3A, C).
Similar to
T. gondii infection, ESP treatment also induced elevation of p-IRE1α, p-PERK, p-eIF2α, cleaved ATF6, and CHOP and attenuation of procaspase-12 in a time-dependent manner (
Fig. 3B, D). Both
T. gondii infection and ESP treatment induced an obvious increase in the BIP protein level at 8 hr and maintained the high level until 24 hr. In contrast,
T. gondii infection reduced the protein levels of Ero1-Lα and PDI at 2 hr, which then slightly recovered, whereas ESP treatment gradually reduced the protein levels of Ero1-Lα and PDI.
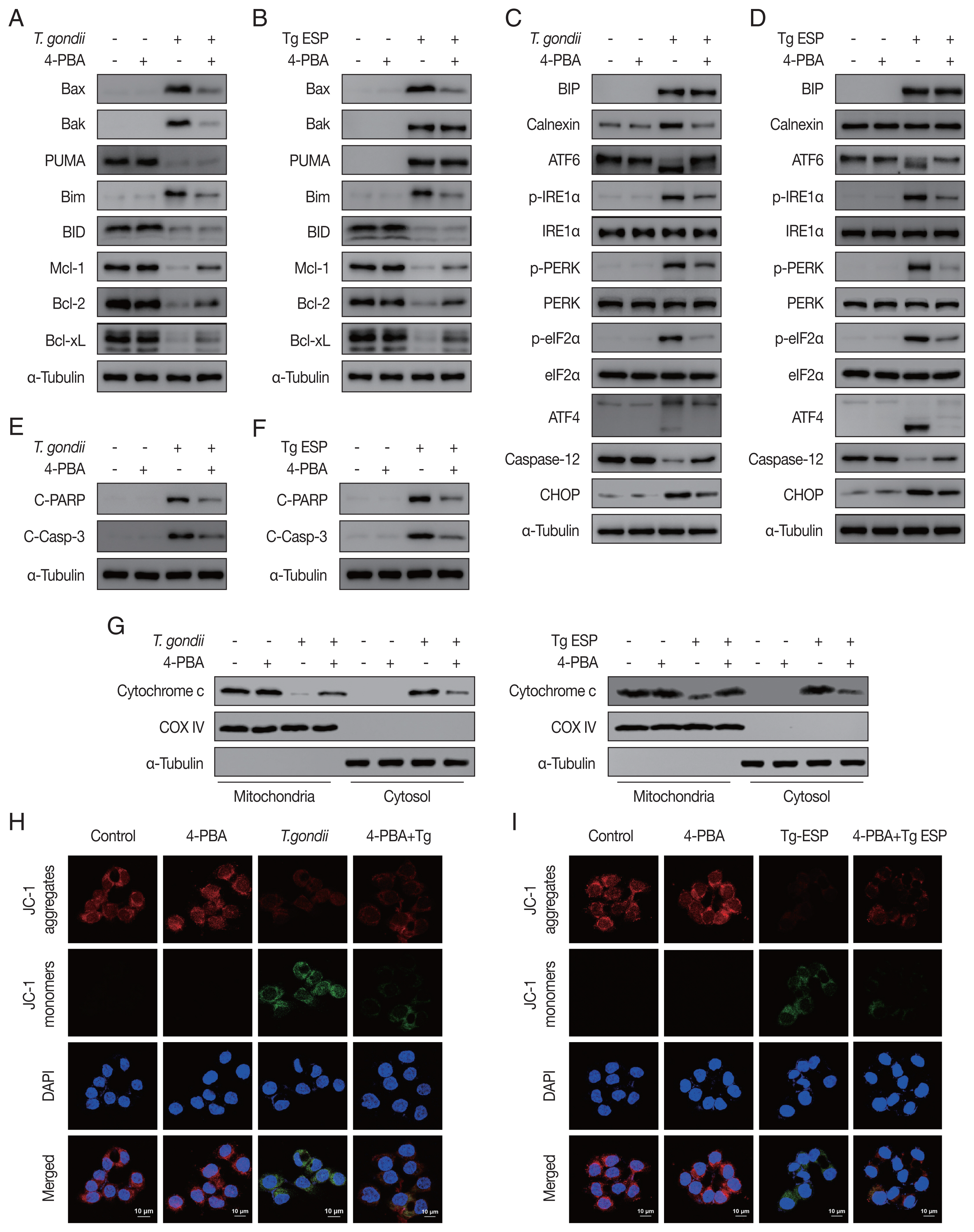
Our results showed that both
T. gondii- and ESP-induced alterations in the expression of Bcl-2 family members (
Fig. 4A, B) and ER stress-related proteins (
Fig. 4C, D) were reversed with 4-PBA pretreatment. In addition, 4-PBA pretreatment attenuated the protein levels of cleaved PARP and cleaved caspase 3 in
T. gondii-infected or ESP-treated FHs 74 Int cells (
Fig. 4E, F).
T. gondii infection- or ESP treatment-derived the release of cytochrome c from the mitochondria into the cytoplasm, which was also prevented by 4-PBA pretreatment (
Fig. 4G and
Supplementary Fig. S1A, B). Confocal microscopy imaging of JC-1 dye staining showed that
T. gondii infection- or ESP treatment-induced MMP depolarization in FHs 74 Int cells was rescued by 4-PBA pretreatment (
Fig. 4H, I and
Supplementary Fig. S1C, D). These results demonstrate that the
T. gondii RH strain and ESP regulated ER stress, which mediates host mitochondrial dysfunction and apoptosis.
DISCUSSION
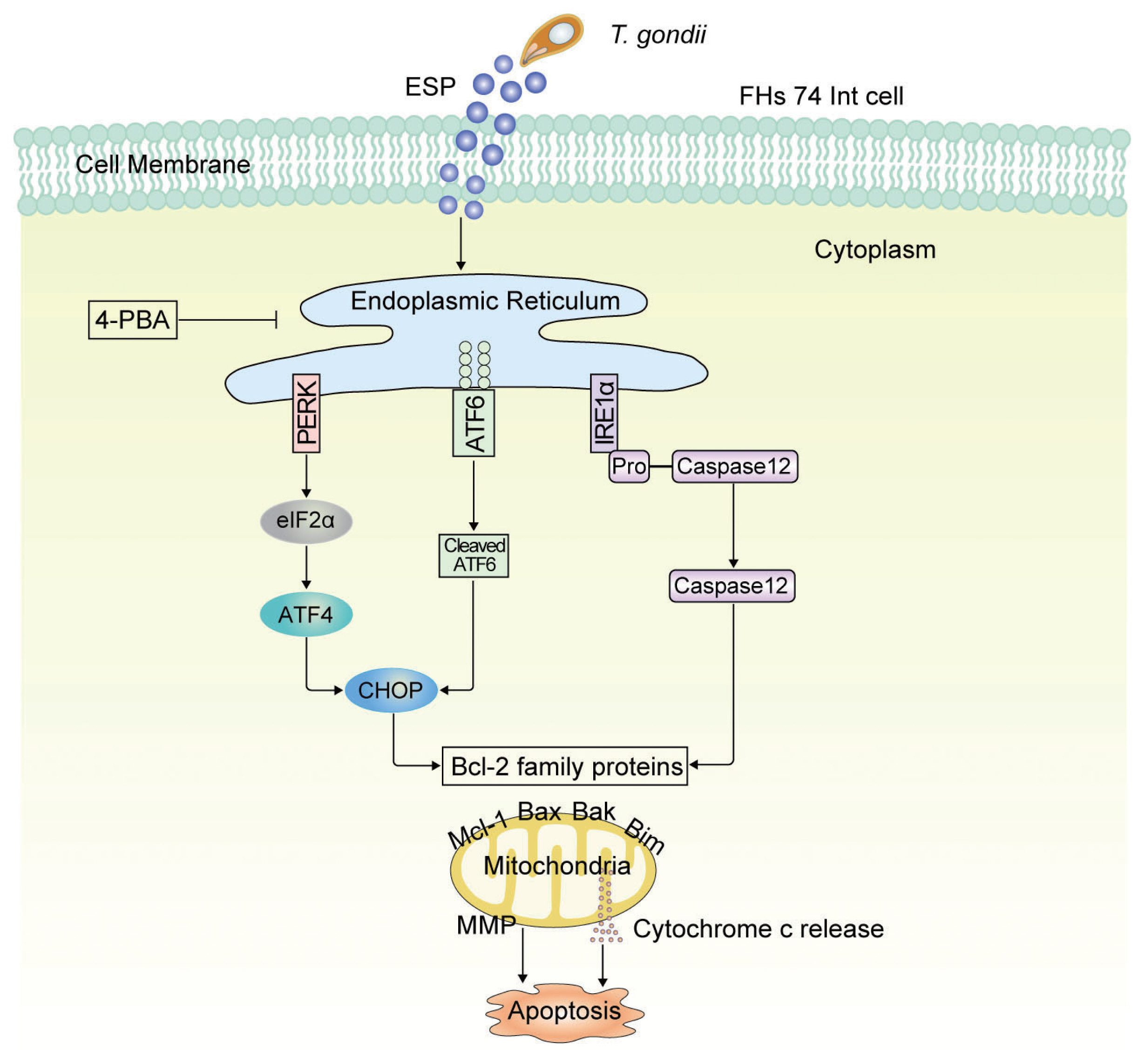
The current study revealed that T. gondii infection and ESP treatment significantly induced cell cytotoxicity and caspase-3-dependent mitochondrial apoptosis in FHs 74 Int cells. In addition, T. gondii infection and ESP treatment dramatically increased Bax, Bak and Bim levels but decreased the Mcl-1 protein level in a time-dependent manner. ER stress induction by T. gondii infection and ESP was confirmed by detecting increases in BIP, calnexin, p-IRE1α, p-PERK, p-eIF2α, CHOP, cleaved ATF6 and cleaved ATF4. Moreover, pretreatment with the ER stress inhibitor 4-PBA attenuated the levels of Bcl-2-, ER stress- and apoptosis-related proteins. More importantly, T. gondii infection and ESP treatment caused a strong decrease in MMP, but 4-PBA pretreatment attenuated MMP depolarization. These data indicate that T. gondii infection and ESP treatment induced apoptosis via the PERK/eIF2α/ATF4/CHOP, ATF6/CHOP, and IRE1α/Caspase-12 ER stress-derived mitochondrial pathways in human FHs 74 Int cells.
T. gondii infection induces NLRP3 inflammasome activation in FHs 74 Int cells [
19]. Notably, the NLRP3 inflammasome can also cause apoptosis in response to a broad spectrum of proapoptotic stimuli [
21,
22]. Hence, we hypothesized that
T. gondii infection may have a functional role in regulating FHs 74 cell apoptosis. The present study found that
T. gondii infection and ESP treatment induced FHs 74 cell cytotoxicity and caspase-dependent apoptosis. Our results are consistent with those of previous reports indicating that
T. gondii infection induces cytotoxicity and apoptosis in cells in a time-dependent manner [
17,
23]. However, our results were contrary to those of Nishikawa et al. [
14], who reported that
T. gondii inhibits ultraviolet light-induced apoptosis through multiple interactions with the mitochondrion-dependent programmed cell death pathway. These different effects on host cell effects may be a result of differences in cell type, treatment method, parasite infection time, and host cell microenvironments.
Bcl-2 plays an important role in maintenance of mitochondrial integrity by preventing apoptosis-induced cytochrome c release, and cytochrome c is involved in activating downstream caspases that trigger apoptosis [
9,
23]. Mitochondria are not only a checkpoint for apoptosis progression but also release toxic proteins controlled by Bcl-2 family members at the mitochondrial outer membrane [
8]. We utilized a JC-1 MitoMP detection kit to detect mitochondrial membrane potential (MMP). Because JC-1 dye exhibits potential-dependent accumulation in mitochondria with a fluorescence shift from green (488 nm) to red (561 nm), mitochondrial depolarization is indicated by a decrease in the red:green fluorescence intensity ratio [
24]. We found that
T. gondii infection and ESP treatment apparently increased Bax, Bak, and Bim protein levels but that BID, Mcl-1, Bcl-2, and Bcl-xL levels were diminished in a time-dependent manner. JC-1 dye staining revealed that
T. gondii infection and ESP treatment caused a strong decrease in MMP, indicated by a reduction in red and increase in green JC-1 fluorescence signals. This event is governed by a complex interplay between Bcl-2 family member proteins [
8,
25].
The endoplasmic reticulum (ER) is an elaborate cellular organelle essential for cell function and survival. Conditions that interfere with ER function lead to accumulation and aggregation of unfolded proteins, which are detected by ER transmembrane receptors that initiate the unfolded protein response (UPR) to restore normal ER function. If ER stress is prolonged or the adaptive response fails, apoptotic cell death ensues [
26].
T. gondii infection time-dependently increased phospho-IRE1α, phospho-PERK, phospho-eIF2α, CHOP, and cleaved-ATF6 protein levels. In addition,
T. gondii ESP treatment also elevated p-IRE1α, p-PERK, p-eIF2α, CHOP, and cleaved ATF6 protein levels. Recently, Augusto et al. [
27] described that
T. gondii infection activates each of the UPR sensor proteins, including IRE1, via ER stress and IRE1 controls host cell migration in infected immune cells
in vitro and also facilitates dissemination of
T. gondii in vivo. The phosphorylation of ATF6β by
T. gondii ROP18 induced its proteosomal degradation and reduction in ATF6β mediated gene expression after induction of UPR. ATF6β-deficient mice exhibit a high susceptibility to infection by the parasite, indicating that ATF6β has a key role in resistance against
T. gondii infection [
28]. Our results show that small intestinal epithelial cell apoptosis being initiated by endoplasmic reticulum stress via activation of caspase-12, CHOP, and the JNK pathway in
T. gondii infection and ESP treatment.
The findings of the present study suggest that ER stress-related protein expression is regulated by
T. gondii infection and ESP treatment and positively correlated with mitochondrial dysfunction in FHs 74 Int cells. Pretreatment with the ER stress inhibitor 4-PBA attenuated the levels of ER stress-, Bcl-2- and apoptosis-related proteins, such as p-IRE1α, p-PERK, p-eIF2α, cleaved ATF6, CHOP, Bax, Bak, Bim, cleaved PARP, and cleaved caspase-3, and MMP depolarization in
T. gondii-infected or ESP-treated FHs 74 Int cells. Notably, the present results indicate that
T. gondii infection and ESP treatment caused Bcl-2 family proteins to regulate mitochondrial apoptosis in FHs 74 Int cells via the PERK/eIF2α/ATF4/CHOP, ATF6/CHOP, and IRE1α/caspase-12 pathways (
Fig. 5). Therefore, ER stress response-regulated intracellular signaling pathways might play crucial roles in small intestinal immunopathology following oral infection with
T. gondii.
Notes
-
The authors declare that they have no conflicts of interest.
Supplementary Information
Supplementary Fig. S1.
Toxoplasma gondii- and ESP-regulated mitochondrial dysfunction is dependent on ER stress in FHs 74 Int cells. FHs 74 Int cells were pretreated with various concentrations of the ER stress inhibitor 4-phenylbutyrate (4-PBA) for 4 hr and subsequently infected with T. gondii at an MOI of 10 (A, C) or treated with 1 μg/ml ESP for 18 hr (B, D). (A, B) Mitochondrial and cytosolic fractions from the cells were extracted and using Western blotting to examine the distribution of Cytochrome c. COX IV and α-Tubulin were used as markers of mitochondria and cytosol, respectively. (C, D) JC-1 staining was observed via confocal microscopy imaging. Bar graph showing the ratio of JC-1 red and green fluorescence indicative of mitochondrial membrane potential. ***P<0.001, compared to the untreated control cells under the same conditions or T. gondii infection or ESP treated control cells.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81771612), the Characteristic Innovation Projects of Guangdong Universities (2018KTSCX081), the Guangdong Basic and Applied Basic Research Foundation (2019A1515011715), and the Competitive Allocation Project of Zhanjiang Municipal Science and Technology Development Special Fund (2020A01021).
Fig. 1
Toxoplasma gondii and ESP induced cytotoxicity and apoptosis in human fetal small intestinal epithelial FHs 74 Int cells. FHs 74 Int cells were stimulated with the T. gondii RH strain at a multiplicity of infection (MOI) of 10 or with 1 μg/ml ESP for 0, 2, 8, and 24 hr. (A) The lactate dehydrogenase (LDH) level in the medium. The horizontal lines in each group represent mean±SD. ***P<0.001, compared to the untreated control cells. (B) Cleaved PARP and cleaved caspase 3 protein levels. (C, D) Expression of cytochrome c of in mitochondrial and cytosolic fractions.
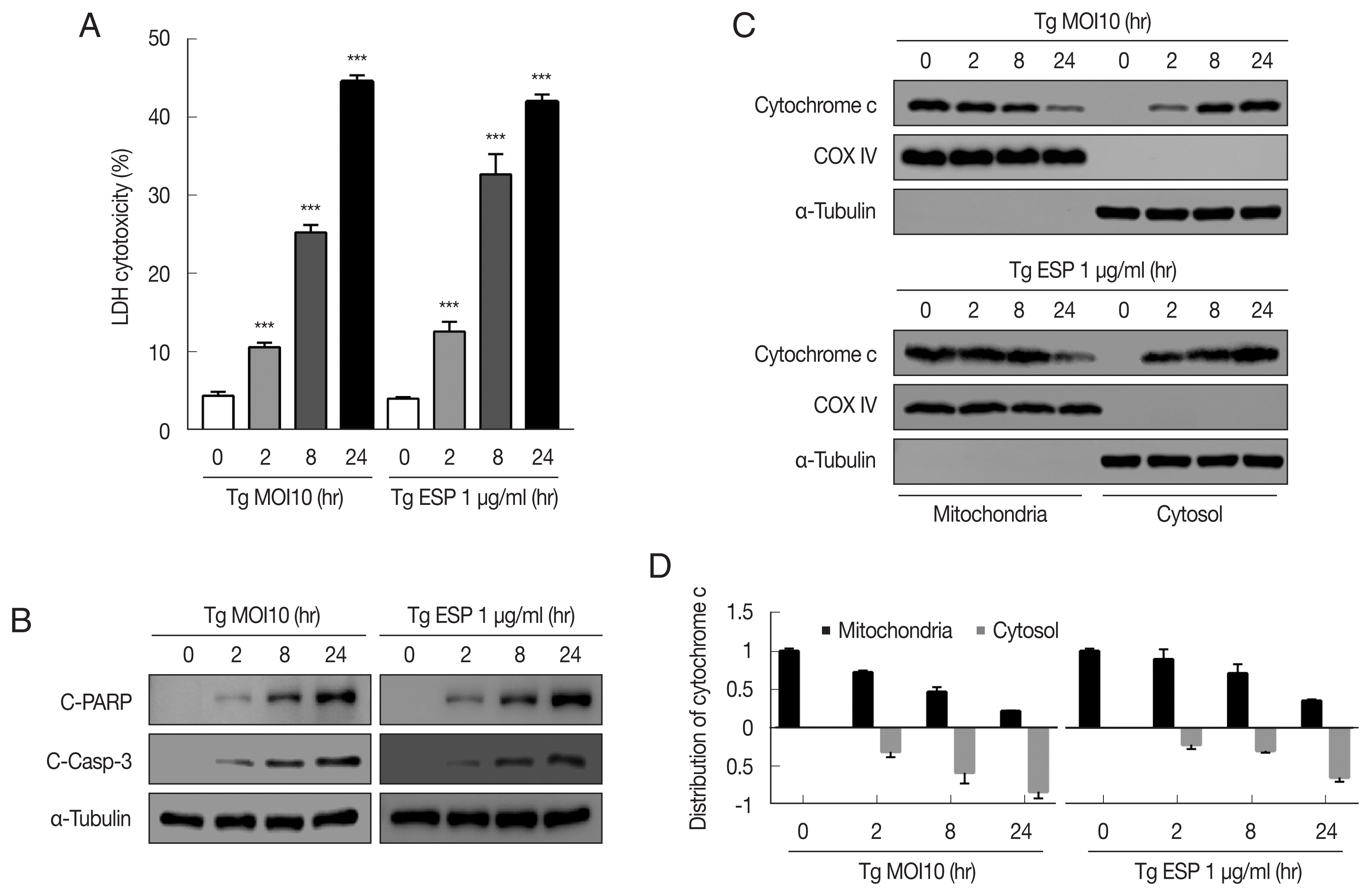
Fig. 2
Toxoplasma gondii and ESP derived mitochondrial apoptosis in FHs 74 Int cells. (A, B) Protein expression of proapoptotic Bcl-2 and prosurvival Bcl-2 family members. (C, D) JC-1 staining shows red fluorescence for high mitochondrial membrane potential, while green fluorescence for low mitochondrial membrane potential. Bar graph showing the ratio of JC-1 red and green fluorescence.
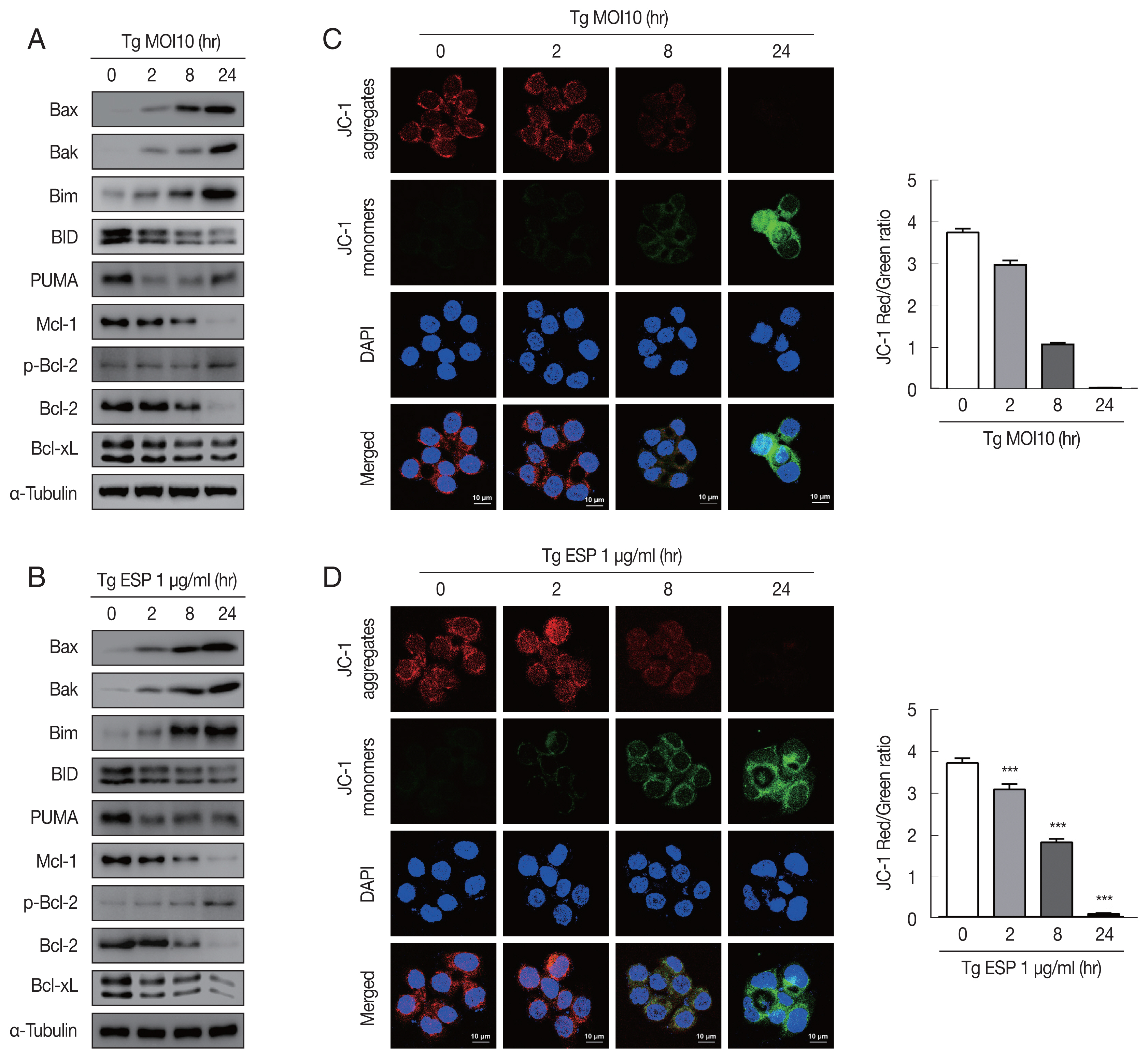
Fig. 3
Toxoplasma gondii and ESP caused ER stress in FHs 74 Int cells. FHs 74 Int cells were stimulated with the T. gondii RH strain at an MOI of 10 (A, C) or with 1 μg/ml ESP for 0, 2, 8, and 24 hr (B, D). Bar plot depicting the indicated proteins to α-tubulin ratios. *P<0.05, **P<0.01, ***P<0.001.
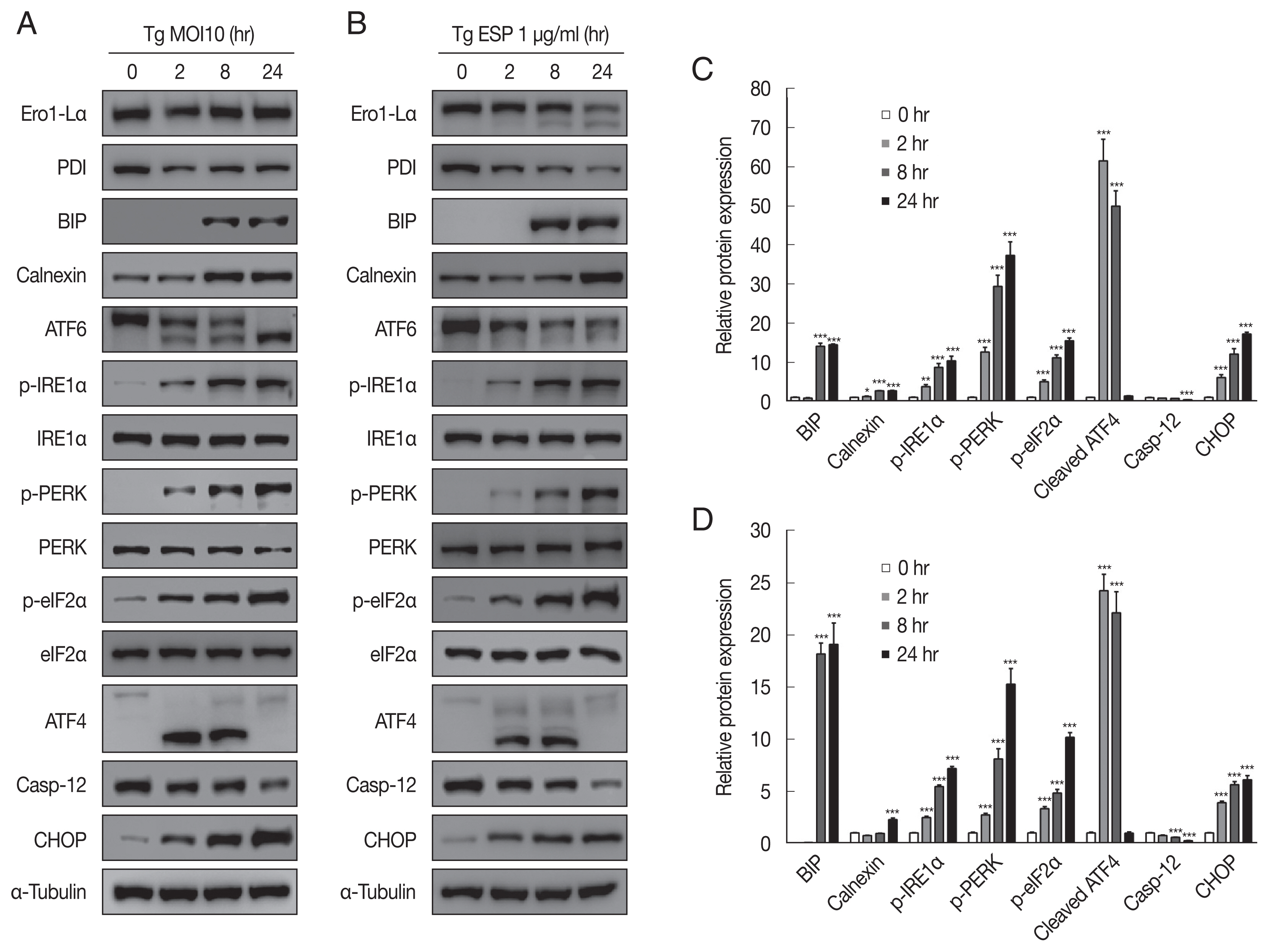
Fig. 4
Toxoplasma gondii- and ESP-motivated mitochondrial apoptosis in FHs 74 Int cells. FHs 74 Int cells were pretreated with various concentrations of the ER stress inhibitor 4-phenylbutyrate (4-PBA) for 4 hr and subsequently infected with T. gondii at an MOI of 10 or treated with 1 μg/ml ESP for 18 hr. (A, B) The Bcl-2 family member. (C, D) ER stress-related protein. (E, F) Cleaved PARP and cleaved caspase 3 protein levels. (G) Mitochondrial and cytosolic fractions analyzed for expression of cytochrome c. (H, I) JC-1 staining observed via confocal microscopy imaging.
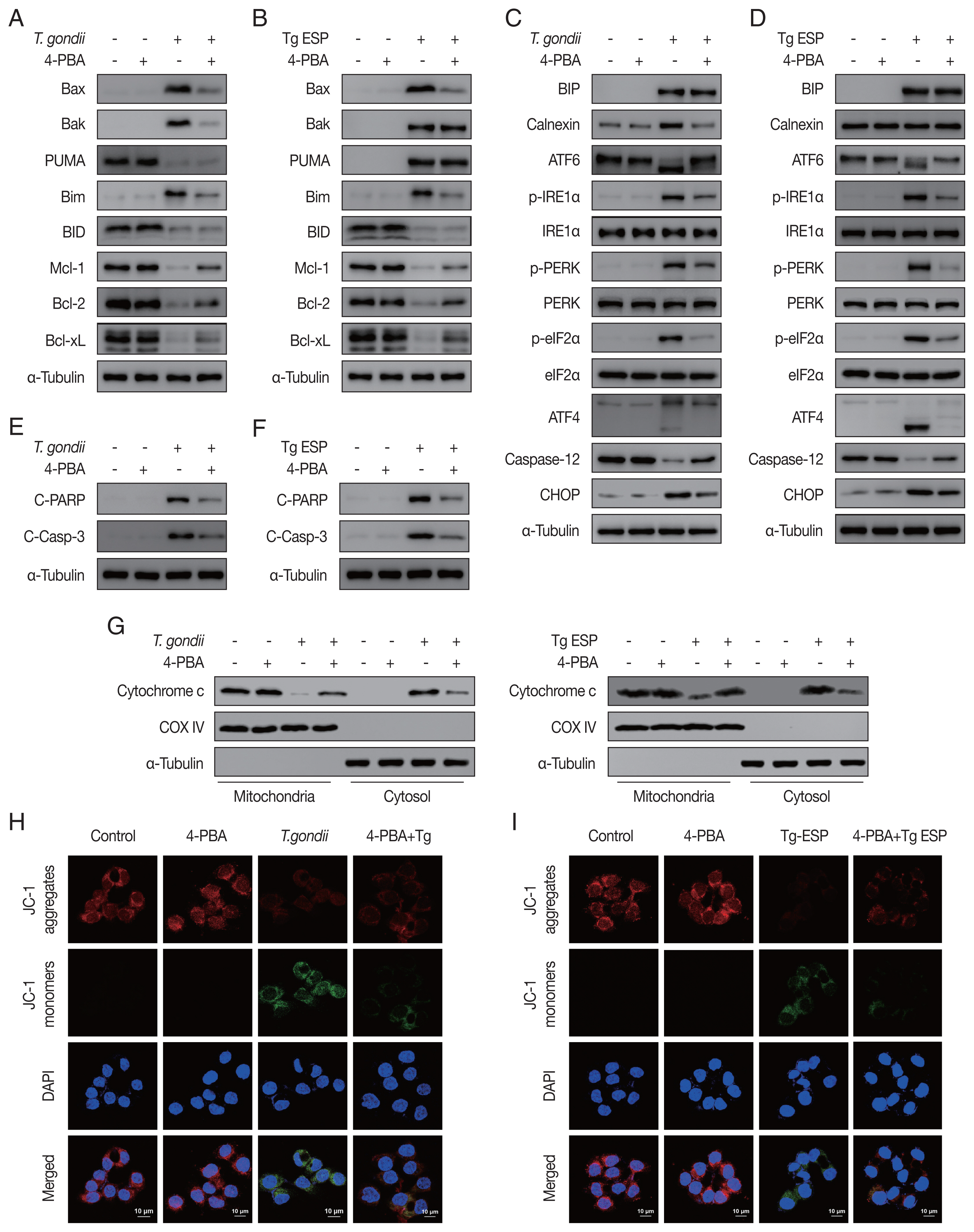
Fig. 5Schematic model of mitochondrial apoptosis induced by T. gondii and ESP stimulation in FHs 74 Int cells. T. gondii and ESP induce ER stress-mediated mitochondrial dysfunction.

References
- 1. Mendez OA, Koshy AA. Toxoplasma gondii: Entry, association, and physiological influence on the central nervous system. PLoS Pathog 2017;13:e1006351. https://doi.org/10.1371/journal.ppat.1006351
- 2. Liu Q, Wang ZD, Huang SY, Zhu XQ. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasit Vectors 2015;8:292. https://doi.org/10.1186/s13071-015-0902-6
- 3. Esch KJ, Petersen CA. Transmission and epidemiology of zoonotic protozoal diseases of companion animals. Clin Microbiol Rev 2013;26:58-85. https://doi.org/10.1128/CMR.00067-12
- 4. Desmonts G, Couvreur J. Congenital toxoplasmosis. A prospective study of 378 pregnancies. N Engl J Med 1974;290:1110-1116. https://doi.org/10.1056/NEJM197405162902003
- 5. Zhou W, Quan JH, Lee YH, Shin DW, Cha GH. Toxoplasma gondii proliferation require down-regulation of host Nox4 expression via activation of PI3 kinase/Akt signaling pathway. PLoS One 2013;8:e66306. https://doi.org/10.1371/journal.pone.0066306
- 6. Pestechian N, Shahreza HK, Faridnia R, Kalani H. Manipulation of IL-10 gene expression by Toxoplasma gondii and its products. Med J Islam Repub Iran 2016;30:410. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5038996/
- 7. Grilo AL, Mantalaris A. Apoptosis: a mammalian cell bioprocessing perspective. Biotechnol Adv 2019;37:459-475. https://doi.org/10.1016/j.biotechadv.2019.02.012
- 8. Bhola PD, Letai A. Mitochondria—judges and executioners of cell death sentences. Mol Cell 2016;61:695-704. https://doi.org/10.1016/j.molcel.2016.02.019
- 9. Mammari N, Halabi MA, Yaacoub S, Chlala H, Dardé ML, Courtioux B. Toxoplasma gondii modulates the host cell responses: an overview of apoptosis pathways. Biomed Res Int 2019;2019:6152489. https://doi.org/10.1155/2019/6152489
- 10. Xu X, He L, Zhang A, Li Q, Hu W, Chen H, Du J, Shen J. Toxoplasma gondii isolate with genotype Chinese 1 triggers trophoblast apoptosis through oxidative stress and mitochondrial dysfunction in mice. Exp Parasitol 2015;154:51-61. https://doi.org/10.1016/j.exppara.2015.04.008
- 11. Contreras-Ochoa CO, Lagunas-Martínez A, Belkind-Gerson J, Díaz-Chávez J, Correa D. Toxoplasma gondii invasion and replication within neonate mouse astrocytes and changes in apoptosis related molecules. Exp Parasitol 2013;134:256-265. https://doi.org/10.1016/j.exppara.2013.03.010
- 12. Quan JH, Cha GH, Zhou W, Chu JQ, Nishikawa Y, Lee YH. Involvement of PI 3 kinase/Akt-dependent Bad phosphorylation in Toxoplasma gondii-mediated inhibition of host cell apoptosis. Exp Parasitol 2013;133:462-471. https://doi.org/10.1016/j.exppara.2013.01.005
- 13. Payne TM, Molestina RE, Sinai AP. Inhibition of caspase activation and a requirement for NF-κB function in the Toxoplasma gondii-mediated blockade of host apoptosis. J Cell Sci 2003;116:4345-4358. https://doi.org/10.1242/jcs.00756
- 14. Carmen JC, Hardi L, Sinai AP. Toxoplasma gondii inhibits ultraviolet light-induced apoptosis through multiple interactions with the mitochondrion-dependent programmed cell death pathway. Cell Microbiol 2006;8:301-315. https://doi.org/10.1111/j.1462-5822.2005.00622.x
- 15. Slee EA, Keogh SA, Martin SJ. Cleavage of BID during cytotoxic drug and UV radiation-induced apoptosis occurs downstream of the point of Bcl-2 action and is catalysed by caspase-3: a potential feedback loop for amplification of apoptosis-associated mitochondrial cytochrome c release. Cell Death Differ 2000;7:556-565. https://doi.org/10.1038/sj.cdd.4400689
- 16. Cao Y, Long J, Liu L, He T, Jiang L, Zhao C, Li Z. A review of endoplasmic reticulum (ER) stress and nanoparticle (NP) exposure. Life Sci 2017;186:33-42. https://doi.org/10.1016/j.lfs.2017.08.003
- 17. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta 2013;1833:3460-3470. https://doi.org/10.1016/j.bbamcr.2013.06.028
- 18. Wang T, Zhou J, Gan X, Wang H, Ding X, Chen L, Wang Y, Du J, Shen J, Yu L. Toxoplasma gondii induce apoptosis of neural stem cells via endoplasmic reticulum stress pathway. Parasitology 2014;141:988-995. https://doi.org/10.1017/S0031182014000183
- 19. Quan JH, Huang R, Wang Z, Huang S, Choi IW, Zhou Y, Lee YH, Chu JQ. P2X7 receptor mediates NLRP3-dependent IL-1β secretion and parasite proliferation in Toxoplasma gondii-infected human small intestinal epithelial cells. Parasit Vectors 2018;11:1. https://doi.org/10.1186/s13071-017-2573-y
- 20. Son ES, Nam HW. Detection and characterization of excretory/secretory proteins from Toxoplasma gondii by monoclonal antibodies. Korean J Parasitol 2001;39:49-56. https://doi.org/10.3347/kjp.2001.39.1.49
- 21. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012;36:401-414. https://doi.org/10.1016/j.immuni.2012.01.009
- 22. Malireddi R, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol 2019;9:406. https://doi.org/10.3389/fcimb.2019.00406
- 23. Graumann K, Schaumburg F, Reubold TF, Hippe D, Eschenburg S, Lüder CG. Toxoplasma gondii inhibits cytochrome c-induced caspase activation in its host cell by interference with holo-apoptosome assembly. Microb Cell 2015;2:150-162. https://doi.org/10.15698/mic2015.05.201
- 24. Quan JH, Gao FF, Ismail HAHA, Yuk JM, Cha GH, Chu JQ, Lee YH. Silver nanoparticle-induced apoptosis in ARPE-19 cells is inhibited by Toxoplasma gondii pre-infection through suppression of NOX4-dependent ROS generation. Int J Nanomedicine 2020;15:3695-3716. https://doi.org/10.2147/IJN.S244785
- 25. Liu N, Tang M. Toxic effects and involved molecular pathways of nanoparticles on cells and subcellular organelles. J Appl Toxicol 2020;40:16-36. https://doi.org/10.1002/jat.3817
- 26. Gorman AM, Healy SJ, Jäger R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther 2012;134:306-316. https://doi.org/10.1016/j.pharmthera.2012.02.003
- 27. Augusto L, Martynowicz J, Amin PH, Alakhras NS, Kaplan MH, Wek RC, Sullivan WJ Jr. Toxoplasma gondii Co-opts the Unfolded Protein Response To Enhance Migration and Dissemination of Infected Host Cells. mBio 2020;11:e00915-00920. https://doi.org/10.1128/mBio.00915-20
- 28. Yamamoto M, Ma JS, Mueller C, Kamiyama N, Saiga H, Kubo E, Kimura T, Okamoto T, Okuyama M, Kayama H, Nagamune K, Takashima S, Matsuura Y, Soldati-Favre D, Takeda K. ATF6β is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J Exp Med 2011;208:1533-1546. https://doi.org/10.1084/jem.20101660

 , Chunchao Li1,†
, Chunchao Li1,†