Signaling Role of NADPH Oxidases in ROS-Dependent Host Cell Death Induced by Pathogenic Entamoeba histolytica
Article information

Abstract
All living organisms are destined to die. Cells, the core of those living creatures, move toward the irresistible direction of death. The question of how to die is critical and is very interesting. There are various types of death in life, including natural death, accidental death, questionable death, suicide, and homicide. The mechanisms and molecules involved in cell death also differ depending on the type of death. The dysenteric amoeba, E. histolytica, designated by the German zoologist Fritz Schaudinn in 1903, has the meaning of tissue lysis; i.e., tissue destroying, in its name. It was initially thought that the amoebae lyse tissue very quickly leading to cell death called necrosis. However, advances in measuring cell death have allowed us to more clearly investigate the various forms of cell death induced by amoeba. Increasing evidence has shown that E. histolytica can cause host cell death through induction of various intracellular signaling pathways. Understanding of the mechanisms and signaling molecules involved in host cell death induced by amoeba can provide new insights on the tissue pathology and parasitism in human amoebiasis. In this review, we emphasized on the signaling role of NADPH oxidases in reactive oxygen species (ROS)-dependent cell death by pathogenic E. histolytica.
INTRODUCTION
Entamoeba histolytica, a tissue-invasive intestinal extracellular protozoan parasite, infects about 50 million people worldwide, and causes more than 40,000 death each year [1]. Infection with E. histolytica is acquired by drinking or ingestion of water or food containing cysts. During infection, amoebic trophozoites induce cell death via apoptosis and necrosis in host tissues, and causes colitis accompanied by severe diarrhea and abdominal pain. It also causes liver abscesses with high fatality. Representative virulence factors of dysentery amoeba include amoebic Gal/GalNAc lectin, cysteine protease (CP), and amoebapore [2]. When attached to the mucin layer of the colon epithelium through Gal/GalNAc lectin present on the surface of the amoeba, CP is secreted to degrade the colonic mucus [3]. During the death process of the colon epithelial cells by amoeba, the pre-interleukin (IL)-1β secreted from the dying or dead cells are cleaved by activated amoebic CP [4], which subsequently mediates IL-1β-dependent activation of NF-kB pathways in the neighboring living cells [5,6]. This biochemical cascade finally leads secretion of inflammatory chemokines including IL-8 [7,8]. Acute inflammatory cells, including neutrophils, migrate to inflamed tissues and secrete several inflammatory mediators that can trigger a larger inflammatory response. On the other hand, apoptosis and engulfment of immune cells by amoebic trophozoites benefits the survival of the amoeba and host tissues, which can induce minimal tissue inflammatory responses [9]. Amoeba can also modulate or destroy immune effector cells by suppressing respiratory burst or nitric oxide production from macrophages [10]. The delicate interactions between the host cells and the amoebae contribute to the regulation and balance of parasitism. Therefore, understanding of the mechanisms involved in host cell death induced by the amoeba can be valuable to provide new insights on parasitism and tissue pathology in humans with infected with amoeba. In this review, we highlight the intracellular signaling molecules leading to cell death by dysenteric E. histolytica.
VARIOUS MODES AND FORMS OF CELL DEATH
The first form of cell death found is necrosis, which is characterized by the destruction of cell membrane, resulting in global cellular swelling and breakdown of intracellular organelles or nuclei [11]. The excessive leakage of cellular substances from the cell to the neighboring tissues causes severe inflammatory reactions [12]. However, a form of cell death that was not explained by necrosis was discovered in 1972 and the term “apoptosis” began to be used to refer to a unique form of cell death. Apoptosis, a programed cell death (PCD), is characterized by nuclear chromatin condensation, cellular shrinkage, expansion of vesicles, and membrane blisters, but mitochondriae are morphologically intact. Apoptosis is distinguished from necrosis. This type of cell death has often been difficult to observe in vivo conditions as dying cells were rapidly engulfed by tissue macrophages. However, advances in cell biological technologies led to the discovery of PCD in the 1990s. PCD has further subdivided into apoptotic cell death (apoptosis) and non-apoptotic cell death (necroptosis, pyroptosis, autophagy, and etc.) [11,13]. Since then, various forms of cell death that do not belong to this category have been found and classified according to the morphological changes in cells. In 2018, the Nomenclature Committee on Cell Death classified cell death into 12 categories, which included necrosis, a non-PCD, and PCDs (apotosis/anoikis, autophagy, ferroptosis, pyroptosis, NETosis, necroptosis, and etc.) [13].
THE UNIQUENESS OF HOST CELL DEATH INDUCED BY E. histolytica
It has been generally accepted that cell death caused by amoeba can occur differently from the common pathways of apoptosis. For example, during the apoptosis in Jurkat T cells by amoeba, the activity of effector caspase (caspase 3) was induced, although the activity of its upstream caspase 9 or caspase 8 was not observed to increase [14]. Dysenteric amoeba easily killed caspase-8 deficient cells treated with caspase-9 inhibitor [14]. Mouse liver cells lacking the Fas/FasL and TNF receptor I (TNFR I) signaling pathways was also killed upon stimulation with amoeba [15]. UV-induced apoptosis was inhibited in Bcl-2 overexpressing cells, but amoeba-induced apoptosis was not inhibited at all [16]. Furthermore, pro-apoptotic mitochondria-derived factors such as Bax or cytochrome c do not appear to be associated with E. histolytica-induced Jurkat T cell death [17]. These collective results suggest that mitochondriae might be bystanders during the cell death caused by E. histolytica.
Electron microscopic (EM) observation of cell death of human neutrophils by amoeba confirmed the predominance of apoptotic cell death, although some necrotic cell death co-existed [18]. A similar morphological characteristics was observed by EM when CaCo2 cells, a colon epithelial cell line, were cocultured with live amoebic trophozoites (Fig. 1). During the cell death process, various cell shapes such as cells with condensed or swollen nuclei were mixed. In addition, images of the amoeba eating the cytoplasm of the host cells and cells phagocytosed by amoeba as a whole were observed. These results suggest that cell death by amoeba can be induced via several different pathways, and that the process of cell death may vary depending on the cell types or sizes. However, the exact signaling molecules involved in various types of cell death induced by amoebae remained largely unveiled.
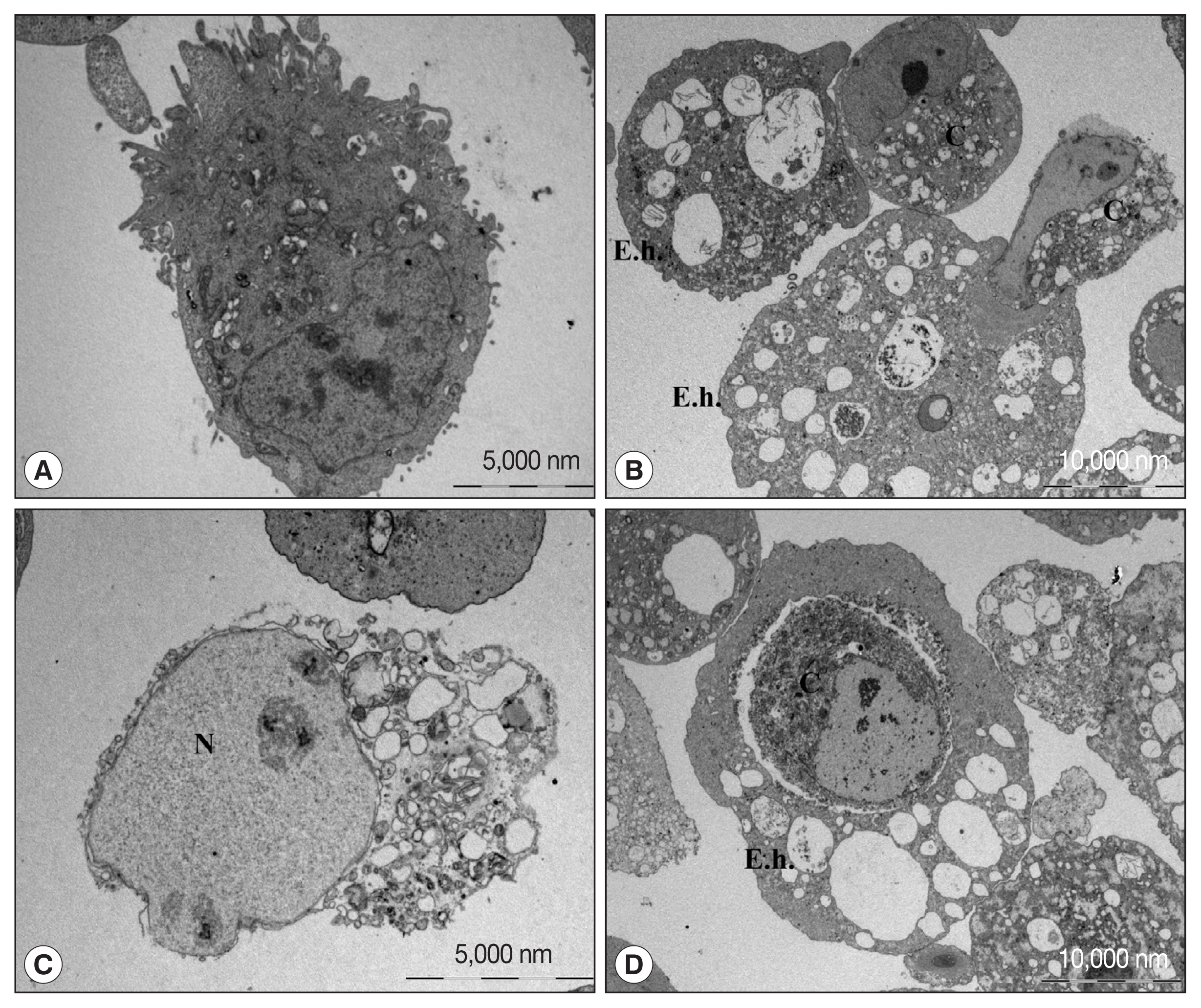
Transmission electron microscopic observation of Caco-2 cells incubated with or without trophozoites of E. histolytica. (A) Caco-2 cells incubated without amoebae have an irregular surface, heterogenous chromatin, and intact mitochondriae. (B–D) Caco-2 cells in contact with amoebae showing chromatin condensation and cell shrinkage. The nucleus of the Caco-2 cells incubated with amoeba is swollen and cytoplasmic contents are disrupted. Caco-2 cells are engulfing or eaten by amoeba. C denotes Caco-2 cells. E. h denotes trohpzoites of Entamoeba histolytica. N denotes nucleus of Caco-2 cells.
It has recently been demonstrated that dysenteric amoeba induces cell death by consuming intact living cells little by little through a process called trogocytosis [19,20]. The trogocytosis process was first reported as a phenomenon that occurs in immune cells, which means ‘nibble’ in the ancient Greek word trogo. Amoebic trogocytosis of host cells was typically observed in live Jurkat T cells [19]. Continuous nibbling leads to loss of the membrane integrity of the Jurket T cells, accelerating in dismantles integrity of the cells [19]. The trogocytosis process by amoeba occurs within 1 min of contact with host cells, and when a living host cell loses its membrane integrity and dies, the amoebae detached from the dead cells without further biting [19,20].
The trogocytosis is a fairly unusual phenomenon that causes host cell death by the amoeba. Interestingly, host cells were eaten by the amoeba by trogocytosis and phagocytosis, depending on the size of the host cells. For example, cells are ingested through phagocytosis alone in the case of small cells such as red blood cells; however, only up to ~20% of the cells are ingest by phagocytosis in the case of relatively large Jurkat cells (approximately 12 μm) and the remaining ~80% of the cells are eaten through trogocytosis [19,20]. It is presumed that the larger the size of the host cell, the higher the induction of host cell death by trogocytosis.
The cell size of the neutrophils we observed approximated 10–18 μm, and the Caco-2 cells were 40–50 μm in their diameters. Due to these apparent differences in cell size, it is postulated that there are more diverse forms of cell death in colon epithelial cells than in neutrophils when encounter with amoebic trophozoites. Amoeba-induced trogocytosis can provide favorable condition that amoebae protect themselves as a mean of evading host s protective immunity by acquiring human cell membrane proteins and labeling them on the amoeba surface [21,22]. Further research is needed on the signaling molecules responsible for trogocytosis-mediated host cell death induced by E. histolytica.
SIGNALING MOLECULES INVOLVED IN NON-ROS-DEPENDENT CELL DEATH INDUCED BY E. histolytica
In 1998, it was discovered that contact with the amoeba irreversibly elevates the intracellular Ca2+ in the host cells. This study also showed that morphologically various forms were found in dead cells co-incubated with amoeba [23]. In 2000, the Huston group first revealed that E. histolytica induced phosphatidylserine (PS) externalization and activation of caspase 3 in Jurkat T cells, showing apoptotic cell death in Jurkat T cells [14]. Parasite-host interaction via amoebic Gal/GalNAc lectin was evidenced to be absolutely necessary for host cell apoptosis. In 2007, the activation of calpain, a Ca2+-dependent CP, has subsequently been reported to play an important role in caspase-3-mediated Jurkat T cell death by E. histolytica. For instance, calpain-dependent calpastatin cleavage can regulate capase-3 activation during apoptosis in Jurkat T cells co-incubuated with pathogenic E. histolytica [24]. In contrast to the findings observed with parasitic amoeba, Jurkat T cells co-incubuated with free-living amoeba Naegleria fowleri, which causes severe tissue inflammation in primary amoebic encephalitis of humans, were found to undergo to die via necroptosis, but not via apoptosis. Interestingly, receptor-interacting protein 1 (RIP) 1-mediated ROS-dependent necroptosis occurs without activation of caspase cascade during Jurkat T cell death induced by pathogenic N. fowleri [25]. These results suggest that the ability of immune cells to actively determine how to die is more advantageous in terms of host defense according to the characteristics of the amoeba introduced into the body.
During amoeba-induced apoptosis of Jurkat T cells, tyrosine dephosphorylation of intracellular proteins occurs, in which the activity of the protein tyrosine phosphatase 1B (PTP1B), Src homology 2 (SH2) domain-containing protein tyrosine phosphatase 1 or 2 (SHP1, or SHP2) has been found to be directly related to calpain-induced cleavage [26,27]. A recent report showed that co-incubation with pathogenic strain of amoeba rapidly reduced protein expression of intracellular O-GlcNAc transferase (OGT), causing overall protein O-deGlcNAcylation in liver tumor cell line (HepG2 cells), which led to global cellular dismantling [28]. Thus, host cells appear to undergo to die in a variety of ways in which unique signaling mechanisms and related molecules are operated when encountered with amoeba.
Next, we describe our recent researches on the signaling roles of NADPH oxidases in ROS-dependent host cell death induced by E. histolytica.
NADPH OXIDASES AND ROS GENERATION IN MAMMALIAN CELLS
ROS sources of eukaryotic cells are mitochondria, arachidonic acid, and NADPH oxidases (NOXs), which is known as respiratory burst oxidase homologs [29]. ROS act as a second messenger and is known to play a very important role in a variety of intracellular signaling processes such as host defense, cell growth, differentiation, and cell death, and increased gene expression associated with inflammatory circumstances [30].
The NOXs, one of the main intracellular generators of ROS, is classified into 7 isoforms, which included NOX1-5, dual oxidase (DUOX) 1, and DUOX2 [31,32]. All NOX isoforms are structured with a dehydrogenase domain that binds a non-covalently linked flavin cofactor and NADPH substrate with a common catalytic core consisting of 6 transmembrane helices chelating 2 hemes [30,31]. The NADPH oxidase complex was typically composed of a transmembrane flavocytochrome b558 consisting of p91phox and p22phox, and 6 or more protein components, including 4 cytosolic components (p47phox, p67phox, p40phox, and small GTP-binding Rac1 or Rac2 proteins). Interestingly, the expression of NOX isoform varies by tissue or cell type, and the cytosolic component, mechanism, and manner of regulation required for the activity of NOX isoforms are slightly different. Six homologs of NOX2, including NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2 have been highly expressed in non-phagocytic cells and play a role in a variety of intracellular processes.
SIGNALING ROLE OF NOXS IN CELL DEATH OF PHAGOCYTES AND NON-PHAGOCYTES INDUCED BY E. histolytica
NOX2-derived ROS in neutrophils
The phagocytic NADPH oxidase, NOX 2, has long been studied, which is the first form of NOX isoforms characterized [32]. NOX2 primarily works on innate immunity, including anti-bacterial defense. In addition, NOX2 has been reported to participate in a diverse physiological process which included angiogenesis or cell death [32]. The association of NOX2-derived ROS in the apoptotic cell death induced by amoeba was demonstrated in human neutrophils [33]. Neutrophil apoptosis induced by live amoebic trophozoites was evidenced by decreases in the amount of CD16 surface expression on the neutrophils. The neutrophil apoptosis was demonstrated by measuring an increase in phosphatidyl serine (PS) externalization on the cell surface using annexin V, as well as an increase in the amount of cells stained with propidium iodide (PI). When neutrophils were pretreated with pan-caspase inhibitor or caspase-3 specific inhibitor, apoptosis of neutrophils by amoeba was significantly inhibited. Amoeba-induced ROS generation and apoptosis in human neutrophils was remarkably inhibited by diphenyleneiodonium (DPI) pretreatment, a NOX inhibitor. However, cells pretreated with mitochondrial inhibitor rotenone or antioxidants such as NAC, catalase, and glutathione had no effect at all. The signaling linkage between caspase 3 activity and ROS production in neutrophil death has not been discovered, which suggests that ROS generation and caspase-3 functions were independently regulated during the process of E. histolytica-induced neutrophil apoptosis. This result further suggests that there are at least 2 independent pro-death channels in human neutrophils for determination of apoptotic death in response to E. histolytica.
NOX1-derived ROS in colon epithelial cells
The NOX1 is known to be structurally and functionally fairly similar to NOX2 [31]. NOX1 is mainly distributed in colon, prostate, uterus, and vascular cells. NOX1 may play a protective role against pathogens in colons, since its expression is uniquely increased in colon epithelial cells [30,31]. In addition, the distribution of NOX1 increases from the ascending to descending colon segments, in parallel with the increased bacterial burden. Previous studies have reported that NOX1-derived ROS are involved in cell death in colon epithelial cells [34]. On the other hand, NOX1 promotes cancer progression by increasing resistance to cell death [35]. We have shown that the important role of NOX1 in the ROS-mediated death of colon epithelial cells by using colon epithelial cell lines such as Caco-2 cells and HT-29 cells, respectively [36,37]. In colon epithelial cell lines cocultured with amoeba, PS externalization, a characteristic feature of apoptotic cell death, was not particularly pronounced, although the proportion of PI-stained colon cells was significantly increased. In addition, amoeba-induced HT29 cell death was not suppressed by pretreatment with pan-caspase inhibitor. These collective results suggest that colon cells incubated with amoebic trophozoites die by a caspase-independent death mechanism. Rather, calpain appear to be closely involved in death of HT29 cells induced by amoeba, since calpain inhibitor or calpain short interfering RNA (siRNA) efficiently prevented E. histolytica-induced cell death [38]. These results suggest that NOX1-drived ROS and calpain may participate in non-apoptotic death in colon epithelial cells, which can lead to severe colitis induced by E. histolytica.
NOX4-derived ROS in T cells
Recent reports have suggested that NOX4 activity perturbs the redox balance in cells, leading to cell survival or death [39,40]. Jurkat T cells are most commonly used to study cell death in E. histolytica infection. We found that NOX4 was highly expressed in resting state Jurkat T cells [41]. Expression of NOX4 in Jurkat T cells was particularly high in areas close to the cytoplasm and nucleus. NOX4 is known to exist not only in the plasma membranes, but also in the endoplasmic reticulum [42] or nuclear membranes [43]. We observed that PS exposure and DNA fragmentation caused by Amoeba in Jurkat cells were significantly reduced by inhibiting NOX4 activity via NOX inhibitor or siRNA NOX4 [41]. These results might indicate that NOX4 is critical for E. histolytica-induced apoptotic death process in Jurkat T cells. Our ongoing experiments suggest that ROS are involved in the process of hepatocyte death induced by amoeba. When hepatic tumor cell line, HepG2 cells were pretreated with a NOX inhibitor prior to incubation with amoeba, release of lactic dehydrogenase (LDH) from the cells were attenuated [unpublished data]. Further researches are required to unveil what important role NOX plays in ROS-dependent cell death of hepatocytes.
CONCLUSION
By exploring the signaling interactions between parasites and host cells, we can gain a broader understanding of the host cell inflammatory response that occurs in human amoebiasis. Amoeba activate diverse host signaling molecules in a very short time to induce irreversible cell death through various mode of PCD. The main signaling molecules involved in host cell death by the amoeba can be different slightly, depending on the types of host cell or cell size. This review focused on signaling role of NADPH oxidases in ROS-dependent cell death in various immune cells induced by E. histolytica. It would be helpful to provide a new insight on the balance between the tissue pathogenesis and parasitism at the signaling levels of the host’s protective immunity.