Morphological Identification and Phylogenetic Analysis of Laelapin Mite Species (Acari: Mesostigmata: Laelapidae) from China
Article information

Abstract
Laelapinae mites are involved in transmission of microbial diseases between wildlife and humans, with an impact on public health. In this study, 5 mite members in the subfamily Laelapinae (laelapin mites; LM) were morphologically identified by light microscopy, and the phylogenetic relationship of LM was analyzed in combination with the sequence information of part of the LM cytochrome oxidase subunit I (cox1) gene. The morphological identification revealed that 5 mites belonged to the genera Laelaps and Haemolaelaps, respectively. Sequence analysis showed that the ratio of non-synonymous mutation rate to synonymous mutation rate of LM was less than 1, indicating that the LM cox1 gene had undergone purifying selection. Phylogenetic analysis showed that the Laelapinae is a monophyletic group. The genera Haemolaelaps and Hyperlaelaps did not separated into distinct clades but clustered together with species of the genus Laelaps. Our morphological and molecular analyses to describe the phylogenetic relationships among different genera and species of Laelapinae provide a reference for the improvement and revision of the LM taxonomy system.
INTRODUCTION
Laelapinae belongs to the family Laelapidae, 65 species in 35 genera have been reported in China [1]. Laelapinae mites are common ectoparasites of small mammals, especially rodents [2]. The basic process of the development of Laelapinae mites (LM) can be divided into 5 stages: egg, larva, first nymph, second nymph, and adult. They complete the majority of their life cycle in the nest of the host [3]. Adult female mites occur mostly on the host, while adult males (and other life stages) remain primarily in the nests of hosts [4]. Some LM are obligate, non-exclusive haematophages [5] (they feed not only on the blood of the host, but also on small arthropods inhabiting the host’s burrows). Most LM are closely related to diseases and belong to the category of medical gamasid mites. Some mites not only cause gamasid dermatitis, but also have the ability to transmit diseases between wild animals and humans, such as Laelaps jettmari and Haemolaelaps glasgowiare, which carry the haemorrhagic fever with renal syndrome (HFRS) virus [6–8].
Accurate identification of species is the basis for phylogenetic research. However, morphological taxonomic identification is difficult because morphological characters vary with age, developmental stage, environment, and other factors [9]. Since the early 21st century, DNA barcoding technology has rapidly developed, providing opportunities for species identification and gradually becoming one of the main trends in mite taxonomy and molecular systematics [10,11]. The mitochondrial genome is the only extra-nuclear genetic information carrier in animals, and it has the characteristics of matrilineal inheritance, small molecular weight, high mutation rate, minimal recombination, and a fast evolutionary rate [12,13]. The mitochondrial cytochrome oxidase subunit I (cox1) gene is the most conserved of the 3 genes encoding cytochrome oxidase. It has been shown to be suitable for addressing phylogenetic questions over a range of taxonomic levels [14]. At present, arthropod ectoparasites of terrestrial hosts have been studied much less than those of helminths and ectoparasites of aquatic animals [15]. Most scholars on LM have focused on the presence or description of species and the detection of pathogens. Thus, the development of molecular biology techniques has allowed the identification not only of morphologically determined species, but also of morphologically nearly identical species [16].
The phylogenetic relationships among the LM remain unclear. In this study, we determined the cox1 gene sequences of 5 LM and analyzed morphological characteristic features.
MATERIALS AND METHODS
Ethics statement
All methods and procedures used in the capture rodent process were in accordance with the guidelines and regulations approved by the Animal Ethics Committees at Dali University. The approval ID is MECDU-201806-11.
Collection of mites
Mite specimens were obtained from the body surfaces of 5 rodent species (Niviventer fulvescens, Eothenomys miletus, Rattus tanezumi, Rattus nitidus, and Callosciurus erythraeus), in Li-jiang City, Yunnan Province, China, in August 2018. The obtained specimens were transported and stored at the Institute of Pathogens and Vectors, Dali University.
Morphological identification
Specimens of mites were mounted individually in Hoyer’s medium using light microscopy for morphological identification. Since the specimens were sufficiently cleared during the tissue lysis stage of DNA extraction, the typical clearing procedures were not necessary. Identification was based on the morphological characteristics of the mites in reference [1].
DNA extraction and polymerase chain reaction (PCR) amplification
Genomic DNA was extracted from individual mite using a DNeasy Blood and Tissue Kit (Qiagen, Valencia, California, USA). The cox1 genes were all amplified by PCR (High Fidelity PCR system, Roche, Mannheim, Germany) with primers of the cox1 gene (Sense: 5′-GGAGGATTTGGAAATTGATTAGTTCC-3′; Anti sense: 5′-CCCGGTAAAATTAAAATATAAACTTC-3′). The PCR cycle conditions for the gene amplification were: 3 min at 94°C, over 40 cycles of 1 min at 94°C, 1 min at 52°C, 1 min at 72°C, and a final extension step of 10 min at 72°C. The PCR products were analyzed by electrophoresis on a 1% agarose gel. A paired approach was used to sequence PCR products (Thermo Fisher Scientific Genome Sequecing Facility, Guangzhou, China).
Data analysis
Raw sequences were assembled and aligned using the Geneious 11.1 software program (Biometer, Auckland, New Zealand) [17]. The DAMBE [18] software is based on genetic distance analyzed sequence base substitution saturation and the detection of phylogenetic signals. The DnaSP 5.0 [19] software analyzed nonsynonymous (Ka) and synonymous (Ks) substitution ratios. Phylogenetic trees were constructed based on Bayesian in Mrbayes 3.2.5 [20]. MrBayes model chosen a total of 1,000,000 generations were run, with sampling every 1,000 generations, while 4 Markov chains were run, discarding the first 25% of generations as burn-in. The constructed phylogenetic trees were viewed and edited using Figtree 1.4.4 [21].
RESULTS
Morphological characteristics
The 5 mites employed in this study were initially identified as Laelaps echidninus (OL780835), Laelaps nuttalli (OL810027), Laelaps fukienensis (OL806574), Laelaps chini (OL806586), and Haemolaelaps traubi (OL810029). They are morphologically distinct (Fig. 1, Table 1).
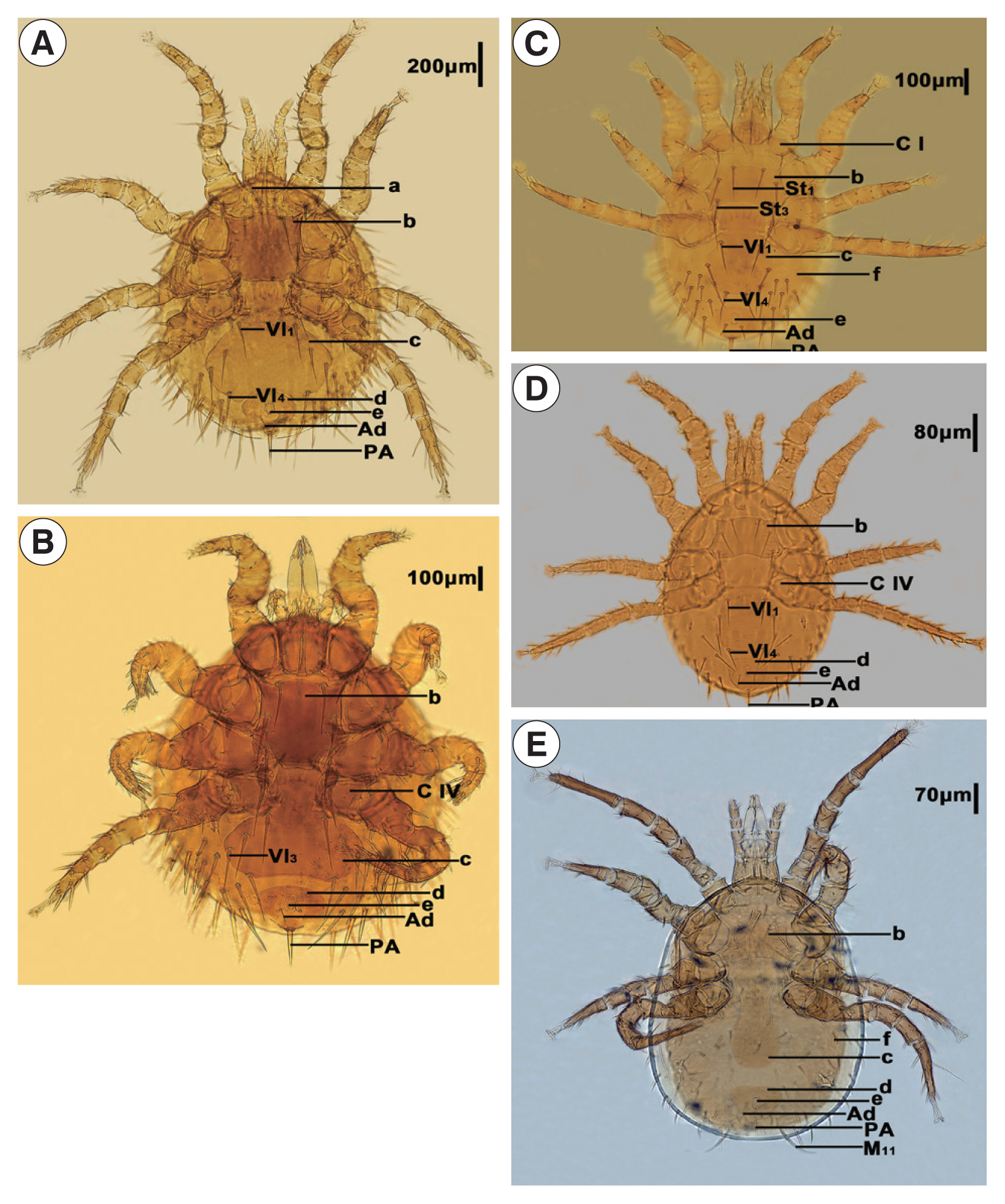
Morphological characteristics of Laelapinae spp. from China. (A) Laelaps echidinus, (B) Laelaps fukiensis, (C) Laelaps chini, (D) Laelaps nuttalli, and (E) Haemolaelaps traubi. a, Tritos ternum; b, Sternal plate; c, Genito-ventral plate; d, anal plate; e, anus; f, metapodal plate; C I, Coxa I; C IV, Coxa IV; Vl1, Genito-ventral plate first pair of setae; Vl3, Genito-ventral plate 3rd pair of setae; Vl4, Genito-ventral plate 4th pair of setae; St1, Sternal plate first pair of setae; St3, sternal plate 3rd pair of setae; Ad, adanal setae; PA, postanal setae.

Identification basis of Laelapinae spp. from China
Sequence analysis
The cox1 gene lengths of 21 mites in 6 genera of Laelapinae, retrieved from GenBank Database (www.ncbi.nlm.nih.gov/) ranged from 418 to 709 bp-long and from 61.0 to 74.6% in AT content (Supplementary Table S1). The sequences of the 5 mites determined in this study ranged from 418 to 440 bp in length; the AT content ranged from 71.4 to 73.2%. The 5 mite sequences were aligned with other Laelapinae mites from GenBank and then trimmed to provide an equivalent sequence. There were 181 conserved sites, 230 variable sites, and 192 parsimony-informative sites, with no base insertions and deletions in the 411 bp alignment of cox1 gene sequences. The non-synonymous mutation rate (Ka) and synonymous mutation rate (Ks) of cox1 sequences were calculated using DnaSP 5.0 software, and the mean value of Ka/Ks was about 0.3.
Phylogenetic analysis based on cox1 gene
To ensure that all sequences were available for phylogenetic analysis, sequence base substitution saturation was analyzed using DAMBE software prior to constructing the phylogenetic tree. A scatter plot was constructed using genetic distance (GTR) as the horizontal coordinate and the number of base conversions and base reversals as the vertical coordinate (Fig. 2). The number of base substitutions in the mt-DNA cox1 gene sequence showed a significant linear relationship with the genetic distance, indicating that it can be used for phylogenetic analysis. Based on the mitochondrial cox1 gene, the phylogenetic tree of Laelapinae mites was constructed by the BI method with Carcinoscorpius rotundicauda (NC019623) as the outgroup (Fig. 3). We observed that the Laelapinae is a monophyletic group, which is divided into 2 main branches (Fig. 3). The first branch consists of 4 species of the genus Tropilaelaps located at the base of phylogenetic tree, and the second branch contains the largest number of species, which consists of 17 species of 5 genera. The 5 species determined in this study are all included in the second branch. Laelaps echidninus (OL780 835), Laelaps nuttalli (OL810027), Laelaps fukienensis (OL806 574) and Laelaps chini (OL806586) are all clustered together with the species of the genus Laelaps. Haemolaelaps traubi (OL810029) does not form a separate clade, but clusters with 2 species (Laelaps chini (OL806586) and Laelaps hilaris (MZ048456)) of the genus Laelaps. The phylogenetic tree constructed by the BI method can summarize the phylogenetic relationship of Laelapinae mites into (Tropilaelaps + (Neocycloplaelaps) + (Laelaps + Hyperlaelaps) + (Haemolaelaps + Laelaps)).

Saturation analysis of base substitutions.

Phylogenetic relationships of the laelapin mites based on cox1 gene.
DISCUSSION
The morphological characters of 5 LM identified by light microscopy were found to be consistent with previous studies [1]. The morphological characters of the 5 mites were distinctly different, and the main discriminatory character to distinguish the genus Laelaps from the genus Haemolaelaps was the number of setae on the genito-ventral plate. According to morphological observations, Laelaps fukienensis (OL806574), Laelaps chini (OL806586), Laelaps echidninus (OL780835), and Laelaps nuttalli (OL810027) conformed to the main morphological characters of the genus Laelaps, i.e., the sternal plate has 3 pairs of setae, the genito-ventral plate has 4 pairs of setae, and the posterior part of the genito-ventral plate is obviously dilated. Haemolaelaps traubi (OL810029) conforms to the main morphological characters of the genus Haemolaelaps, i.e., the sternal plate is wider than long, the genito-ventral plate has only one pair of setae, and M11 of Haemolaelaps traubi (OL810 029) is particularly long as an important character for the identification of this species. Taxonomic identification of the mites is mainly based on traditional morphological characters [11]. LMs are diverse, and many morphological characters are highly variable and indistinguishable in specific environments, while morphological similarities and ambiguous characters occur within the same genus. Therefore, the identification of LM by traditional morphological methods alone has some limitations. In order to get more objective results, we used both morphological and molecular evidence to identify 5 LM. This establishes a foundation for future research into the intraspecific phylogenetic relationships of LM.
In general, synonymous mutations are not subject to natural selection, whereas non synonymous mutations are subject to the action of natural selection. In evolutionary analysis, when Ka/Ks=1, natural selection is considered; when Ka/Ks <1, there might be a negative or purifying selection effect; and when Ka/Ks >1, it is considered that there is a positive or directional selection effect [22]. In the present study, an average Ka/Ks ratio of 0.3 was found for 21 mites of Laelapinae, indicating that the Laelapinae mites experienced a purifying selection effect. This means that the cox1 gene has evolved slowly and is highly conserved in the Laelapinae mites, making it suitable for exploring species taxonomy and phylogenetic relationships.
When we constructed the phylogenetic tree of Laelapinae employing the mitochondrial cox1 gene sequences, we found that Laelapinae is a monophyletic group, with a relatively clear phylogenetic relationship. The species of the same genus always get together preferentially, with high node support rates, indicating that the intra-genus difference is less than the inter-genus difference. The genus Tropilaelaps was located at the base of phylogenetic tree, showing that the genus Tropilaelaps is an older taxon in the Laelapinae. Laelaps fukienensis (OL806574), Laelaps chini (OL806586), Laelaps echidninus (OL780835), and Laelaps nuttalli (OL810027) are well clustered with other species of the genus Laelaps and have a high node support rates. This result indicates that the molecular taxonomy of these 4 species is completely consistent with the traditional morphological classification. Since the genera Haemolaelaps and Hyperlaelaps each involve only one species, namely Haemolaelaps traubi (OL810029) and Hyperlaelaps microti (MZ048468), they cluster with the species of the genus Laelaps and do not form a separate cluster group. This observation differs from the existing taxonomic system of Laelapinae mites and requires further elaboration. The parasite depends primarily on the host for survival. Previous studies have reported that the phylogenetic relationship of the parasite is affected by the phylogenetic relationship of the parasite’s host [23,24]. Therefore, Haemolaelaps traubi (OL810029) and Hyperlaelaps microti (MZ048468) with 4 mites of the genus Laelaps (Laelaps chini (OL806586), Laelaps hilaris (MZ048456), Laelaps kochi (MG414008), and Laelaps schatzi (MK716211)) clustered separately, which may be related to the relatively close kinship of the parasitic hosts of these species. Secondly, the genera Haemolaelaps and Hyperlaelaps may show similar morphological characteristics to the genus Laelaps (for example, the dorsal plate setae are not very long or short, the chelicerae are thick, and the sternal plate has 3 pairs of setae) [1]. In composing the phylogenetic tree, the species of the genera Haemolaelaps and Hyperlaelaps preferentially aggregate with the species of the genus Laelaps. While the genus Hyperlaelaps had been originally proposed as a subgenus of the genus Laelaps [25], many researches did not follow this conclusion. Close clustering of species from different genera has occurred in studies on the phylogenetic relationships of the sesarmid crab, leading to the realignment of related species and the establishment of new genera [26]. The phenomenon of close clustering of species of different genera (Haemolaelaps and Laelaps, Hyperlaelaps and Laelaps) has also been observed in Laelapinae. Based on this, identity of the species of the genera Haemolaelaps and Hyperlaelaps may need to be redefined.Can the genera Hyperlaelaps and Haemolaelaps be considered subgenera of the genus Laelaps, or can Haemolaelaps traubi (OL810029) and Hyperlaelaps microti (MZ048468) be considered members of the genus Laelaps? The validation of this hypothesis will require more in-depth and comprehensive morphological and molecular studies related to more species of the 2 genera.
At present, the phylogeny of LM has been relatively little studied, and the delineation of its taxonomic status and phylogenetic status need to be further refined. Due to the limited amount of information contained in the cox1 gene fragment utilized in this study, it may still be insufficient in resolving the affinities of some species, and the external morphological characters of some species are less different and still diverge in their taxonomic and evolutionary status. It is necessary to investigate more gene sequences as well as more species numbers to further define the taxonomic status and phylogenetic relationships of LM by combining morphological characters.
Supplementary Information
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China [No. 32060143 and 31660314 to W Dong]. The data that support the findings of this study are available in the National Center for Biotechnology Information (NCBI) at [https://www.ncbi.nlm.nih.gov/], reference number [OL806574, OL806586, OL780835, OL810027, OL810029]. We thank Ting Chen for assistance with an initial phylogenetic tree analysis
Notes
The authors declare that they have no conflicts of interests