In Vitro Evaluation of Two Novel Antimalarial Derivatives of SKM13: SKM13-MeO and SKM13-F
Article information

Abstract
Antimalarial drugs play an important role in the control and treatment of malaria, a deadly disease caused by the protozoan parasite Plasmodium spp. The development of novel antimalarial agents effective against drug-resistant malarial parasites is urgently needed. The novel derivatives, SKM13-MeO and SKM13-F, were designed based on an SKM13 template by replacing the phenyl group with electron-donating (-OMe) or electron-withdrawing groups (-F), respectively, to reverse the electron density. A colorimetric assay was used to quantify cytotoxicity, and in vitro inhibition assays were performed on 3 different blood stages (ring, trophozoite, and schizonts) of P. falciparum 3D7 and the ring/mixed stage of D6 strain after synchronization. The in vitro cytotoxicity analysis showed that 2 new SKM13 derivatives reduced the cytotoxicity of the SKM13 template. SKM13 maintained the IC50 at the ring and trophozoite stages but not at the schizont stage. The IC50 values for both the trophozoite stage of P. falciparum 3D7 and ring/mixed stages of D6 demonstrated that 2 SKM13 derivatives had decreased antimalarial efficacy, particularly for the SKM13-F derivative. SKM13 may be comparably effective in ring and trophozoite, and electron-donating groups (-OMe) may be better maintain the antimalarial activity than electron-withdrawing groups (-F) in SKM13 modification.
INTRODUCTION
Malaria is a serious and deadly human disease caused by Plasmodium spp. that are transmitted to humans via the bites of infected female Anopheles mosquitoes. In 2020, there were 241 million cases of malaria, which resulted in 627,000 deaths. In particular, children under the age of 5, pregnant women, patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome (HIV/AIDS), immunocompromised people, mobile populations, and travelers are high risk group [1]. Control, repulsion, and eradication of malaria require a combination of many processes, including preventive measures, vector control, and most importantly, the use of effective antimalarial drugs [1].
As malarial symptoms mainly manifest in the blood stage, this stage is often considered the main target of antimalarial drugs [2]. Identifying compounds or drugs that target different steps in the blood-stage replication process or other life cycle stages may aid develop new therapeutics for the treatment and prevention. However, the efficacy of many drugs in inhibiting different stages of red blood cell (RBC) infection remains unclear [3].
For the prevention and treatment of malaria, chemotherapy can involve either the use of a single drug or a combination of drugs, such as artemisinin-based combination therapy (ATC), which is currently the best available malarial treatment, particularly for Plasmodium falciparum infections [1]. However, recent emergence and spread of malaria resistant to artemisinin derivatives and partner drugs in Southeast Asia have led to the failure of ACT in the greater Mekong sub-region [4]. Safe and well-tolerated next-generation antimalarial drugs targeting parasites at the blood stage are required.
In our previous study, SKM13, a novel derivative, was designed based on a chloroquine (CQ) structural template with a modified side chain, including α, β-unsaturated amides and phenylmethyl groups [5], while we did not test the stage specificity for SKM13. This study tested the stage-specific antimalarial efficacy of the SKM13 template and its two derivatives.
Understanding the stage specificity of the drug is important from several perspectives. The differences between each drug acting at different step/stage may be due attributable to the unique and large differences in the protein expression profile at each stage of the parasite infection in the blood [6–9]. Detailed stage-specificity studies enhance our understanding of how drugs affect parasite replication and clearance [3].
Modification of the phenylmethyl group in SKM13, with either an electron-withdrawing group (-F) or an electron-donating group (-OMe), was conducted to generate 2 new derivatives, SKM13-F and SKM13-MeO. These 2 groups represent the activation and deactivation groups of the electrophilic aromatic substitution, which may influence the antimalarial activity of the original SKM13. We determined the in vitro antimalarial activities of SKM13 and the new derivatives at different blood stages of parasite infection.
MATERIALS AND METHODS
Ethical approval
Korean Red Cross Blood Services provided concentrated human red blood cells for this study using a consent form specifically approved by the exempt review of the Institutional Review Board (IRB) of Seoul National University (IRB No., 2105-005-1205). The experimental methodology used in this study was approved by the Institutional Biosafety Committee of Seoul National University (IBC No., SNUIBC-R211205-2).
Materials
Antimalarial drugs, including CQ and atovaquone, were used as control and acquired from Sigma-Aldrich (St. Louis, Missouri, USA). Propidium iodide and SYTOX Green solution were purchased from BD Pharmingen (San Diego, California, USA) and Life Technologies (Grand Island, New York, USA), respectively. The Differential Quik III Stain Kit was purchased from Polysciences, Inc. (Valley Road, Warrington, Pennsylvania, USA).
CQ-sensitive strains (P. falciparum 3D7 (American Type Culture Collection, ATCC PRA-405D) and P. falciparum D6 (ATCC MRA-285)) and Vero cells were purchased from ATCC (Manassas, Virginia, USA).
Synthesis of SKM13-MeO and SKM13-F
SKM13-MeO and SKM13-F were synthesized using unnatural amino acids after several modifications of our previously successful antimalarial compound, SKM13 [5]. The synthesis process is shown in Supplementary Fig. S1A and described in the Methods section of the Supplementary Information. The purity of the reaction products and compound structures was monitored and confirmed using proton NMR spectroscopy.
Cytotoxicity assay (CC50) in mammalian cells
Vero cells were cultured in a monolayer in a 96-well plate using Dulbecco’s modified Eagle’s medium supplemented with 2% fetal bovine serum for 12 h. Then, they were incubated with serially diluted antimalarial compounds for 48 h. After the incubation period, 20 μl of CellTiter 96 AQueous One Solution reagent (Promega, Hollow Road Madison, Wisconsin, USA) was added to each well and incubated at 37°C in a humidifier with 5% CO2 for 2 h. Absorbance at 490 nm was measured using a Tecan Infinite 200 Pro microplate reader (Tecan Group Ltd., Canton of Zürich, Switzerland).
In vitro culture of P. falciparum
The blood stage of the P. falciparum was propagated in 5% hematocrit of O+ human RBCs suspended in RPMI 1640 with 25 mM HEPES and L-glutamine and supplemented with 1% AlbuMAX I Lipid-Rich BSA, 25 mM sodium bicarbonate, 25 μg/ml gentamycin, 0.05% hypoxanthine, and 1% penicillin. The cultures were maintained at 37°C under 5% O2, 5% CO2, and 90% N2. The medium was changed daily and the number of parasites in the blood was monitored by Giemsa staining of thin methanol-fixed blood smears.
In vitro synchronization of P. falciparum
To synchronize P. falciparum, asexual parasites were treated with 5% D-sorbitol (Sigma-Aldrich) to enrich ring stages. Parasite cultures were centrifuged for 5 min at 500×g. Sterilized and pre-warmed 5% D-sorbitol was added to 10 volumes of the pellet. The cultures were shaken every 2 min with inversion. After 10 min, the cultures were washed twice with RPMI 1640 containing 25 mM HEPES, L-glutamine, and 1% penicillin. The pellet was resuspended in the culture medium to a final hematocrit of 5% for maintenance. The procedure was repeated at least 3 times every 48 h.
Fluorescence-activated cell sorting (FACS)
In vitro antimalarial activity experiments were performed as described previously [5]. Briefly, parasites were seeded in 48-well cell culture plates at a density of 0.5% in 2% hematocrit. The antimalarial control drugs CQ, SKM13, and 2 novel compounds were serially diluted in a malaria culture medium and incubated with the parasites. After incubation, the RBC pellet was stained with SYTOX Green solution. Parasite-infected erythrocyte populations were determined using a BD FACSCanto II flow cytometer (BD Biosciences, Carlsbad, California, USA). The antimalarial activity of the test drugs was determined by the half-maximal inhibitory concentration on malaria growth (IC50).
Statistical analysis
All data were analyzed using GraphPad Prism software (version 9.0; GraphPad Software, San Diego, California, USA). All experimental results are expressed as the mean±standard deviation (SD) of 3 biological repeats.
RESULTS
Synthesis and characterization of 2 new antimalarial drugs derived from SKM13
Two final products, SKM13-MeO and SKM13-F, were prepared from commercially available unnatural amino acids. These are synthetic derivatives of SKM13, which exhibited the best antimalarial effect in our previous study. The phenyl group that replaces the methyl group in chloroquine is large. To improve the antimalarial effect, a substituent was introduced on the phenyl group, facilitating the synthesis of substituted phenyl derivatives using unnatural amino acids.
The procedures for the synthesis of 2 final products were identical, as shown in the scheme in Supplementary Fig. S1A. The amino groups of the unnatural amino acids were protected by the Boc group using excess Boc anhydride under basic conditions to obtain quantitative yields of N-Boc-protected acids (Supplementary Fig. S1A (2a–b)). The conversion of carboxylic acids to N-methoxy-N-methyl amides (Weinreb amides) (Supplementary Fig. S1A (3a and 3b)) was accomplished using dicyclohexylcarbodiimide coupling. After deprotection of the N-Boc group under 5% Trifluoroacetic acid in Dichloromethane, the salts (Supplementary Fig. S1A (4a, 4b)) of amines by TFA were directly used in palladium-mediated coupling reaction with 4,7-dichloroquinoline under the reaction conditions, palladium acetate, cesium carbonate, and BINAP, producing quinolinyl Weinreb’s amides (Supplementary Fig. S1A (5a, 5b)) in moderate yields. Finally, SKM13-F and SKM13-MeO were synthesized by 2 sequential procedures: 1) conversion of Weinreb’s amide to aldehyde by DIBAL-H and 2) Emmons-Horner-Wadsworth olefin synthesis of aldehydes with N-dimethylaminoethyl phosphonate amide using a strong base, otassium tert-butoxide. Purification of the final products was challenging because of the similar polarity of the phosphonate amide and the products, which was overcome by reducing the phosphonate amide. The final structure of each novel compound compared with that of SKM13 is shown in Supplementary Fig. S1B. SKM13 and 2 derivatives had one difference in their chemical structures; the side chains of SKM13-F and SKM13-MeO carried fluorophenyl and methoxyphenyl, respectively.
In vitro antimalarial activity
The cytotoxicity of each compound was assessed by inoculating the drug into Vero cells for 48 h, and the results were evaluated as half-cell cytotoxic concentrations (CC50). CC50 values for CQ and SKM13 were consistent with our previous report [5]. However, SKM13-F and SKM13-MeO had lower CC50 than SKM13 (P<0.001), indicating that 2 novel compounds were more cytotoxic than SKM13 (Fig. 1).
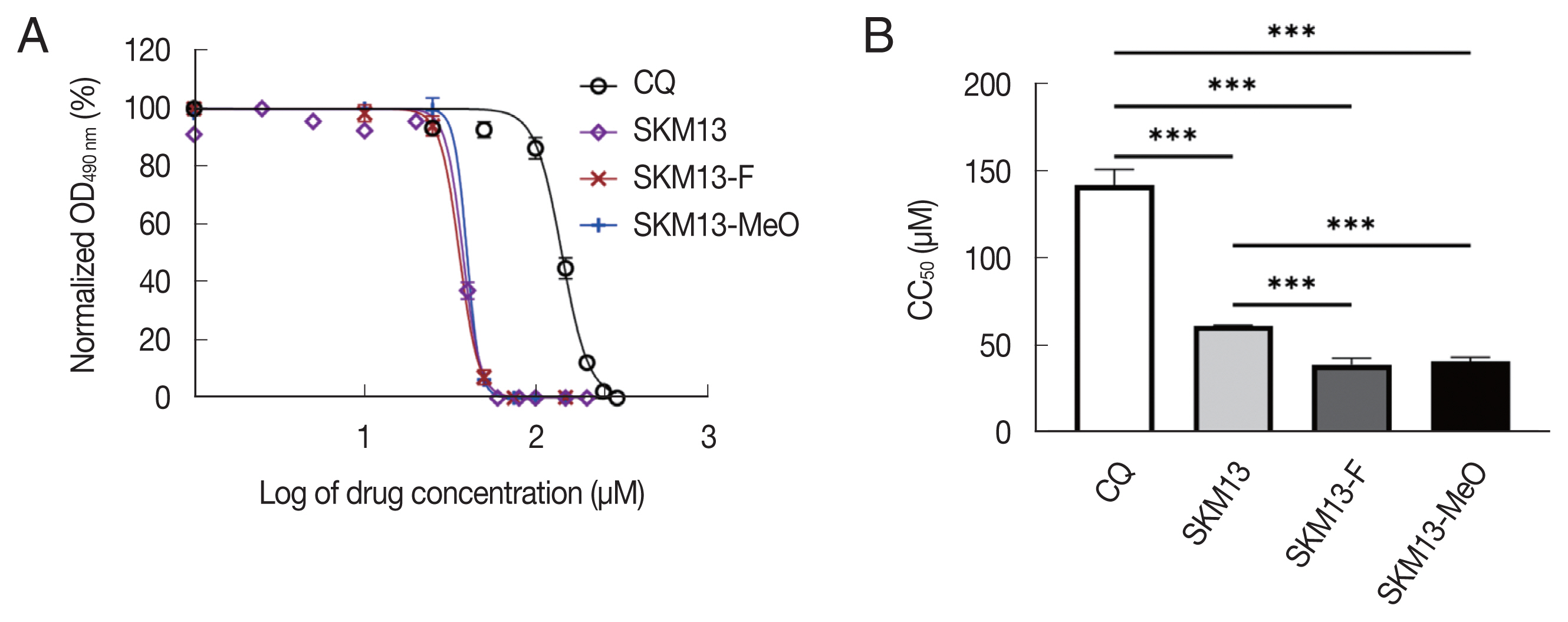
SKM13 deivatives increase toxicity in mammalian cells. (A) The dose-response curve of SKM13 and its novel derivatives, SKM13-F and SKM13-MeO, in comparison with chloroquine (CQ) (B) The summary of the CC50 value for each drug and statistical analysis. (***P<0.001).
We previously reported the IC50 of P. falciparum (3D7) in the mixed stage [5]. The IC50 of the SKM13 series were measured against different developmental stages (ring, trophozoite, and schizont) of P. falciparum. First, malarial parasites were maintained and synchronized to achieve the same stage of parasites using in vitro culture (Supplementary Fig. S2). P. falciparum strains (3D7 and D6) were chosen to determine the antimalarial efficacy of SKM13 and its derivatives (SKM13-MeO and SKM13-F) by measuring the half-maximal concentration of growth inhibition (IC50).
SKM13 and its derivatives showed different antimalarial activities depending on the P. falciparum strains. SKM13 showed IC50 values of 30.6±1, 29.2±1.6, and 52.7±0.4 (mean±SD) at the ring, trophozoite, and schizont stages, respectively, indicating that it was more effective at the ring and trophozoite stages than at the schizont stage (Fig. 2A-C). SKM13-MeO and -F showed higher IC50 values than SKM13 at all 3 stages, implying that SKM13 was more effective than 2 novel derivatives at inhibiting all stages of P. falciparum 3D7. The final SI (CC50/IC50) showed an approximately 2-folds lower value for 2 derivatives of SKM13 than SKM13 itself, suggesting that 2 novel derivatives are less effective than SKM13. There was no difference in SI values between 2 novel SKM13 derivatives.
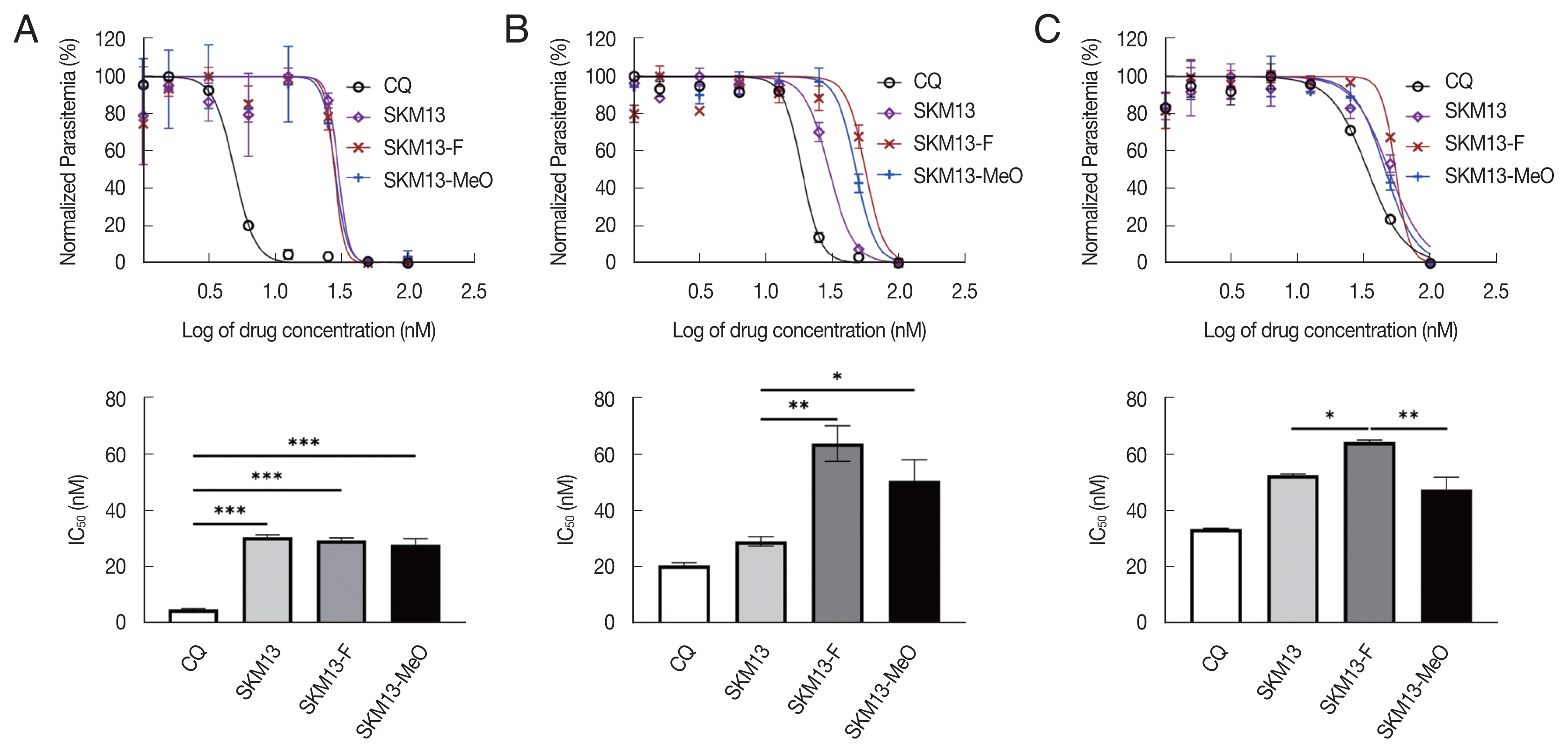
Decrease the antimalarial activity of the SKM13 derivatives in Plasmodium falciparum (3D7) at trophozoite and schizont stages of parasite. The dose-response curve of each drug at ring-stage (A), trophozoite-stage (B), and schizont-stage (C) parasites after treatment with different drug concentrations. (*P<0.05, **P<0.01, ***P<0.001).
D6 strain data suggests that SKM13 might suppress the ring stage better than the other stages because the IC50 value (mean±SD) of SKM13 was increased up to 2 folds in the mixed stage from 11.3±0.2 to 28.5±0.4 (Fig. 3A, B). However, the IC50 of SKM13 was still significantly lower than that of CQ (P<0.001) in D6. SKM13-MeO had a significantly different IC50 than SKM13-F (P<0.001), indicating that the electron-donating group would be more efficient in suppressing P. falciparum than the electron-withdrawing group because CC50 was comparable between 2 modifications. All results of CC50 and IC50 are shown in Table 1.
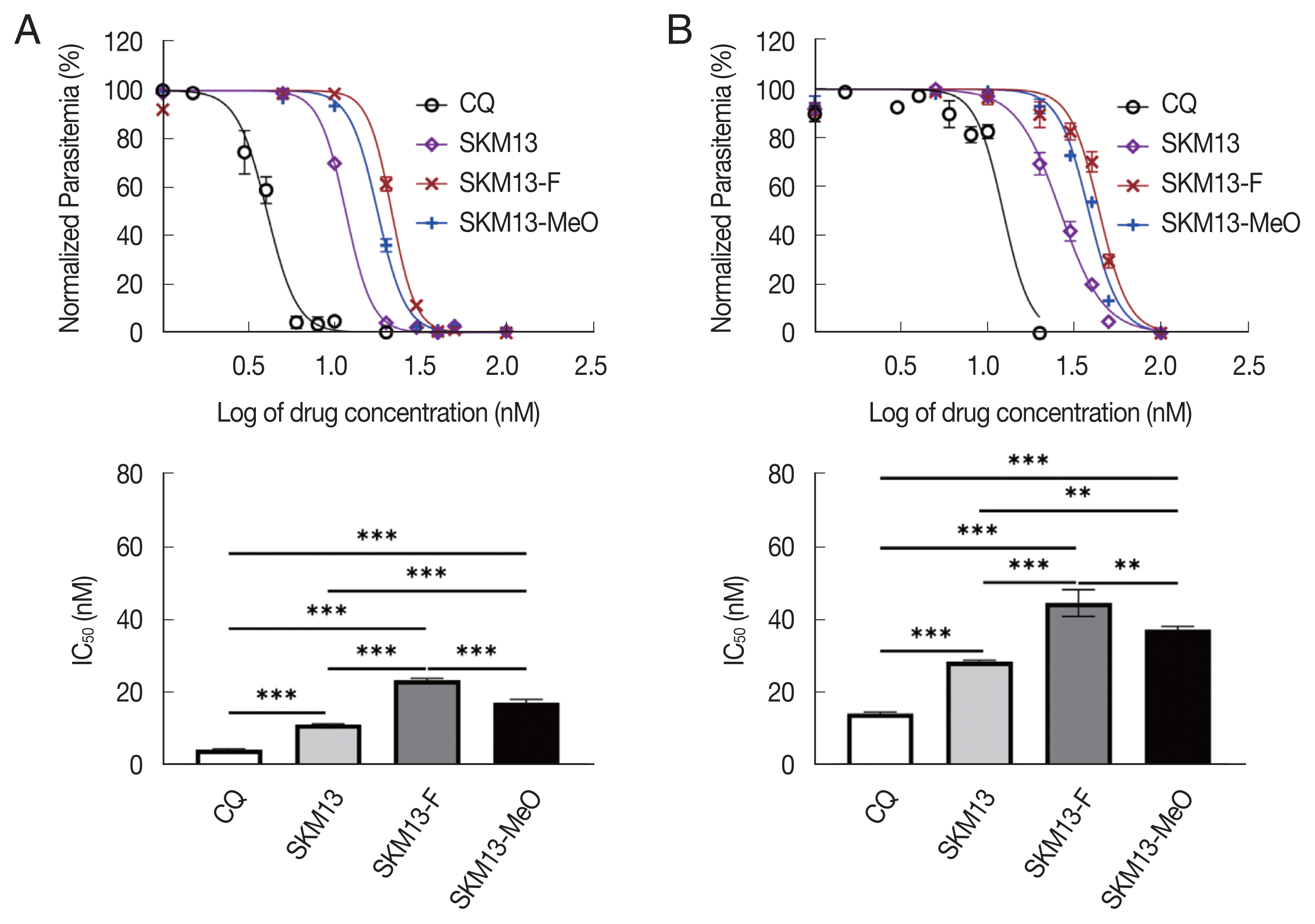
Lower antimalarial efficacy of novel derivatives to SKM13 in Plasmodium falciparum D6 strain. The haft-inhibition concentration (IC50) of CQ, SKM13, SKM13-F, and SKM13-MeO with P. f D6 at synchronized ring stage (A) and mix stage (B). (**P<0.01, ***P<0.001).

Summary of in vitro antimalarial activity
DISCUSSION
Drug resistance has rendered many of the antimalarial compounds ineffective and increasing evidence for artemisinin-resistant malaria requires an urgent need for new antimalarial compounds [10]. P. falciparum transcriptome demonstrated that the strictly controlled progression of parasites through their intra-erythrocytic schizogony is accompanied by a continuous cascade of gene expression, implicating protein diversity according to ring, trophozoite, and schizont stage [11]. A drug that allows the late-stage parasite population to develop normally may not be ideal for treating acute diseases in which rapid clearance is required, and there is a large population of sequestered late-stage parasites. Therefore, if a novel drug targets the ring stage, it would have a great impact on the inhibition of P. falciparum.
The SKM13 was previously considered a promising chemical compound [5], while we could not define the effect of SKM13 on different stages of P. falciparum. In this study, we determined that SKM13 consistently maintained IC50 in ring and trophozoites but not in schizonts. In addition, the newly modified SKM13 derivatives of SKM13 did not have improved IC50.
To investigate the effect of the electron density on the additional phenyl group in SKM13, 2 derivatives, SKM13-F and SKM13-MeO, were synthesized. The fluoro group is a strong electron-withdrawing group that can reduce the reaction rate. In contrast, the methoxy group is an electron-donating group, which can activate the rate of the reaction. Therefore, in this study, the opposite nature of these 2 groups was expected to generate an interesting result regarding antimalarial activities, following the well-known principle of electrophilic aromatic substitution [12]. In our study, SKM13-MeO, with an electron-donating group, had a lower IC50 than SKM-F, corresponding to the well-known theory that fluoro groups might reduce the reaction. Aminoquinolone-related compounds (CQ, AQ, MQ, halofantrine, QN, lumefantrine, and piperaquine) show an expected drug response; parasite death during the trophozoite stage, but no effect on schizont rupture [3]. This may be consistent with our SKM13 derivatives based on the CQ motif because SKM13 and its derivatives did not show any difference in IC50 for the schizont stage. Malaria transmission is dependent on the formation of gametocytes in the human blood, and the sexual conversion rate is determined by the ring stage, implicating that ring stage-specific drugs may be preferred for drug development to efficiently block transmission [13].
Our study has some limitations, including the absence of in vitro results derived from CQ-resistant strain-infected RBCs and in vivo animal models. The study of CQ-resistant strain-infected data will help to interpret the efficacy of 2 derivatives, SKM13-F and SKM13-MeO.
In conclusion, SKM13 derivatives (SKM13-F and SKM13-MeO) did not alter the cell cytotoxicity of SKM13. The inhibitory effect of 2 derivatives against P. falciparum at different developmental stages was not as efficient as that of SKM13 itself. Taken together, the electron-donating group (-OMe) would still be a better contender than the electron-withdrawing group to increase effectiveness against different antimalarial stages.
Supplementary Information
ACKNOWLEDGMENTS
Plasmodium falciparum D6 (ATCC MRA-285) was obtained from BEI Resources, NIAID, NIH: Plasmodium falciparum, Strain D6, MRA-285, contributed by Dennis E. Kyle. Plasmodium falciparum 3D7 was obtained from BEI Resources, NIAID, NIH: Plasmodium falciparum, Strain 3D7 (GL Clone), MRA-1001, contributed by Megan G. Dowler.
This work was supported by the New Faculty Startup Fund of Seoul National University (800-20200549).
Notes
The authors have declared that no conflict of interest exists.