Abstract
The genus Babesia includes parasites that can induce human and animal babesiosis, which are common in tropical and subtropical regions of the world. The gut microbiota has not been examined in hamsters infected by Babesia duncani. Red blood cells infected with B. duncani were injected into hamsters through intraperitoneal route. To evaluate the changes in gut microbiota, DNAs were extracted from small intestinal contents, acquired from hamsters during disease development. Then, the V4 region of the 16S rRNA gene of bacteria was sequenced using the Illumina sequencing platform. Gut microbiota alternation and composition were assessed according to the sequencing data, which were clustered with >97.0% sequence similarity to create amplicon sequence variants (ASVs). Bacteroidetes and Firmicutes were made up of the major components of the gut microbiota in all samples. The abundance of Bacteroidetes elevated after B. duncani infection than the B. duncani-free group, while Firmicutes and Desulfobacterota declined. Alpha diversity analysis demonstrated that the shown ASVs were substantially decreased in the highest parasitemia group than B. duncani-free and lower parasitemia groups. Potential biomarkers were discovered by Linear discriminant analysis Effect Size (LEfSe) analysis, which demonstrated that several bacterial families (including Muribaculaceae, Desulfovibrionaceae, Oscillospiraceae, Helicobacteraceae, Clostridia UGG014, Desulfovibrionaceae, and Lachnospiraceae) were potential biomarkers in B. duncani-infected hamsters. This research demonstrated that B. duncani infectious can modify the gut microbiota of hamsters.
-
Key words: Babesia duncani, gut microbiota, human babesiosis, biomarker, 16S rRNA gene
Introduction
Human babesiosis is induced by apicomplexan tick-transmitted haemoparasites, which are obligate intraerythrocytic protozoa of the genus
Babesia. Among over 100
Babesia species, very minimal is found to induce human disease [
1]. Since the first human case of human babesiosis was found in 1957, 6
Babesia species have been determined as agents responsible for human disease, including
B. microti, B. divergens, B. duncani, B. venatorum, B. motasi, B. crassa [
2–
5]. Recently, an increasing number of
Babesia species (such as
Babesia sp. XXB/HangZhou,
Babesia sp. KO1, and
Babesia microti-like) and cases of human babesiosis have been discovered globally [
6,
7]. Among these species,
B. duncani which was a highly virulent agent comparted with other
Babesia spp. was first reported in a 41-year-old man in Washington State in 1993 [
8]. Apart from America,
B. duncani which caused over 1,000 human cases was also discovered in Canada [
9].
Regarding the apicomplexa parasites, host gut microbiota composition has helped to prevent/treat diseases, such as malaria [
10]. Malaria infections alter the gut microbiota and intestinal tract to influence disease progression. According to previous research, malaria can induce variation in the host immune response and intestinal microbiota changes, which may control the pathogenesis of infectious diseases [
11].
Plasmodium berghei ANKA infectious can cause distinction in the gut microbiota in C57BL/6 and BALB/c mice. When this pathogen was removed, the composition of gut microbiota in infected mice was reconstructed [
12]. Gut microbiota also
aided B. microti transstadial transmission from the nymph to the adult
Haemaphysalis longicornis [
13].
Whether the B. duncani infectious can induce gut microbiota changes have not been explored. We propose that B. duncani infectious may impact the gut microbiota. In this research, we evaluated gut microbiota composition by assessing the B. duncani-free hamsters and B. duncani-infected hamsters to determine potential biomarkers, which gives insight into the association between gut microbiota and babesiosis.
Materials and Methods
Ethics statement
The collection and manipulation of small intestinal contents were approved by the Animal Ethics Committee of the Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences. All procedures were conducted according to the Ethical Procedures and Guidelines of the People’s Republic of China. The study is reported following ARRIVE guidelines.
Hamsters and infection with Babesia duncani strain
Twelve six-week-old male LVG Golden Syrian Hamsters that were bought from Chales River (Beijing, China) were randomly classified into 3 groups (T0, T1, and T2) and kept under pathogen-free conditions. The hamsters were given an ultraviolet-illuminated diet and distilled water at 25°C. Babesia duncani WA1 was procured from ATCC (PRA-302) and kindly gifted by Lanzhou Veterinary Research Institute. Blood samples with 1×106 infected red blood cells were injected intraperitoneally into the T1 and T2 groups hamsters, except for the control group (T0), which were incubated 1×106 hamster red blood cells free-B. duncani infection. When the parasitemia attained 2±0.4% (T1, post-infected day 4), and 20±3.5% (T2, post-infected day 7). The intestine was snipped at the ileocecal junction for collection of the intestinal contents, immediately flash-frozen in liquid nitrogen and stored at −80°C till application. Four free-B. duncani infection hamsters were sacrificed, and contents of the small intestine were collected.
DNA extraction, PCR amplification, and sequencing
DNA extractions from intestinal contents were conducted using Cetyl Trimethyl Ammonium Bromide method, which is adequate for the extraction and purification of DNA from plants and plants derived food staff and is especially suitable for the elimination of polysaccharides and polyphenolic compounds. DNA concentration and purity were identified with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), which was diluted to 1 ng/μl with sterile water. 16S rRNA V4 region was amplified using a set of primers (515F and 806R). PCR amplification was conducted in a total of 50 μl reaction volume containing 25 μl of Phusion High-Fidelity PCR Master Mix (New England Biolabs, USA), 0.5 μm of each primer, and 10 ng templet DNA. The parameters of the PCR reaction included: initial denaturation at 98°C for 1 min, 30 cycles of denaturation at 98°C for the 30 sec, 50°C for 30 sec, and extension at 72°C for 90 sec, with a final extension at 72°C for 5 min. The PCR products were purified with a Qiagen Gel Extraction Kit (Qiagen, Hilden, Germany).
Sequencing libraries were prepared with NEBNext Ultra IIDNA Library Prep Kit (E7645, New England Biolabs, USA) following the manufacturer’s instructions. The library quality was assessed by the Qubit2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. 250 bp paired-end reads were sequenced against libraries with the Illumina NovaSeq platform.
Sequence analysis and species annotation
Sequenced reads were assigned to individual samples/groups based on the barcodes, which together with primer sequences were eliminated. A FLASH package (Ver1.2.11,
http://ccb.jhu.edu/software/FLASH/) was used to integrate paired-end reads with a minimum overlap of 20 base pairs, named raw tags. Low-quality raw tags were filtered using fastp software (Ver0.20.0) to collect clear tags. Following this, chimera sequences were eliminated by comparing clean tags with the reference database (
https://www.arb-silva.de/) using Vsearch (Ver2.15.0) to generate effective tags. Denoise of effective tags was conducted with deblur module in the QIIME2 software (QIIME2-202006) to generate amplicon sequence variants (ASVs), of which with abundance less than 5 were exempted. Species annotation was conducted using QIIME2 software and the Silva database [
14].
The phylogenetic association of each ASV and the different species among groups/samples were identified using QIIME2 software, which was also employed for multiple sequence alignments and normalization of the absolute abundance of ASVs. Normalized ASVs were employed to evaluate alpha diversity, based on 7 indices (observed ASVs, Chao1, Shannon, Simpson, Dominance, Good coverage, and Pielou). Among these indices, Observed ASVs, Chao1, and Dominance were chosen to assess community richness, while Shannon and Simpson were used to determining community diversity. Furthermore, Good’s coverage has deemed the parameter of sequencing depth; Pielou was employed to assess the species evenness.
Beta-diversity was identified based on the ASVs to assess the diversity of bacterial communities among samples and groups (T0, T1, and T2). Beta-diversity was evaluated by calculating weighted and unweighted UniFrac distances that show the distance between each pair of samples. Principle coordinate analysis (PCoA) was conducted using weighted and unweighted UniFrac distances to visualize the ordination and clustering among the samples.
QIIME2 was employed to determine multiple species in samples/groups and considerably different species were identified using MetaStat and T-test analysis. Biomarkers were evaluated using Linear discriminant analysis Effect Size (LEfSe) software (Version 1.0, LDA score threshold is 4).
Availability of data and materials
Results
Confirmation of B. duncani infection
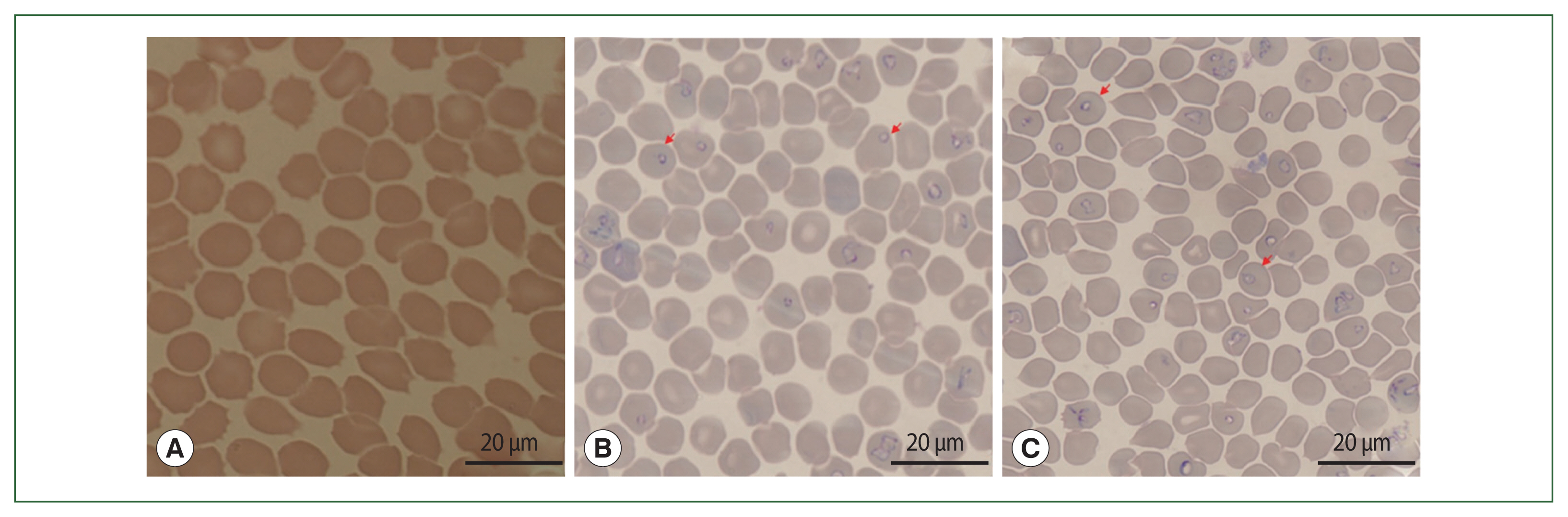
On day 0, hamsters in T1 and T2 were infected with
B. duncani via intraperitoneal injection. Two thousand red blood cells were counted and parasitemia was estimated as the following formula: infected RBC/total number of counted RBC×100%. The parasitemia of infected hamsters belonging to T1 and T2 reached 1±0.2% (on the 4th day) and 10±1.3% (on 8th day), respectively (
Fig. 1). On 9th day after infection with blood samples bearing
B. duncani, as experimental hamsters began to die, the collection of intestinal contents was discontinued.
We obtained 12 intestinal contents from hamsters (4 control hamsters and 8 hamsters infected by
B. duncani), sent to extract DNA and sequence. A total of 1,621,884 raw paired-end reads were derived from the samples. For each sample, the number of sequences ranged from 99,346 to 116,424, with an average number of 108,126±4,726 (
Table 1). After integrating the paired-end sequence, filtering low-quality base/reads, and removing chimeras, we finally acquired a total of 1,036,148 high-quality sequences, which ranged from 36,587 to 78,769 per sample, with an average of 69,077±9,772. The effectiveness that was very low in all samples was assessed by identifying the percentage of effective tags in raw paired-end reads. The values of effectiveness in all samples ranged from 32.5 to 72.1%. The GC content ranged from 53 to 54.7% in all samples, with an average of 53.9%. A total of 2,197 microbial ASVs were determined with 97% of sequence similarity. The average number of ASVs in each sample was 618. In general, the ASV number was almost comparable between the T0 (1,306) and T1 (1,426) groups (
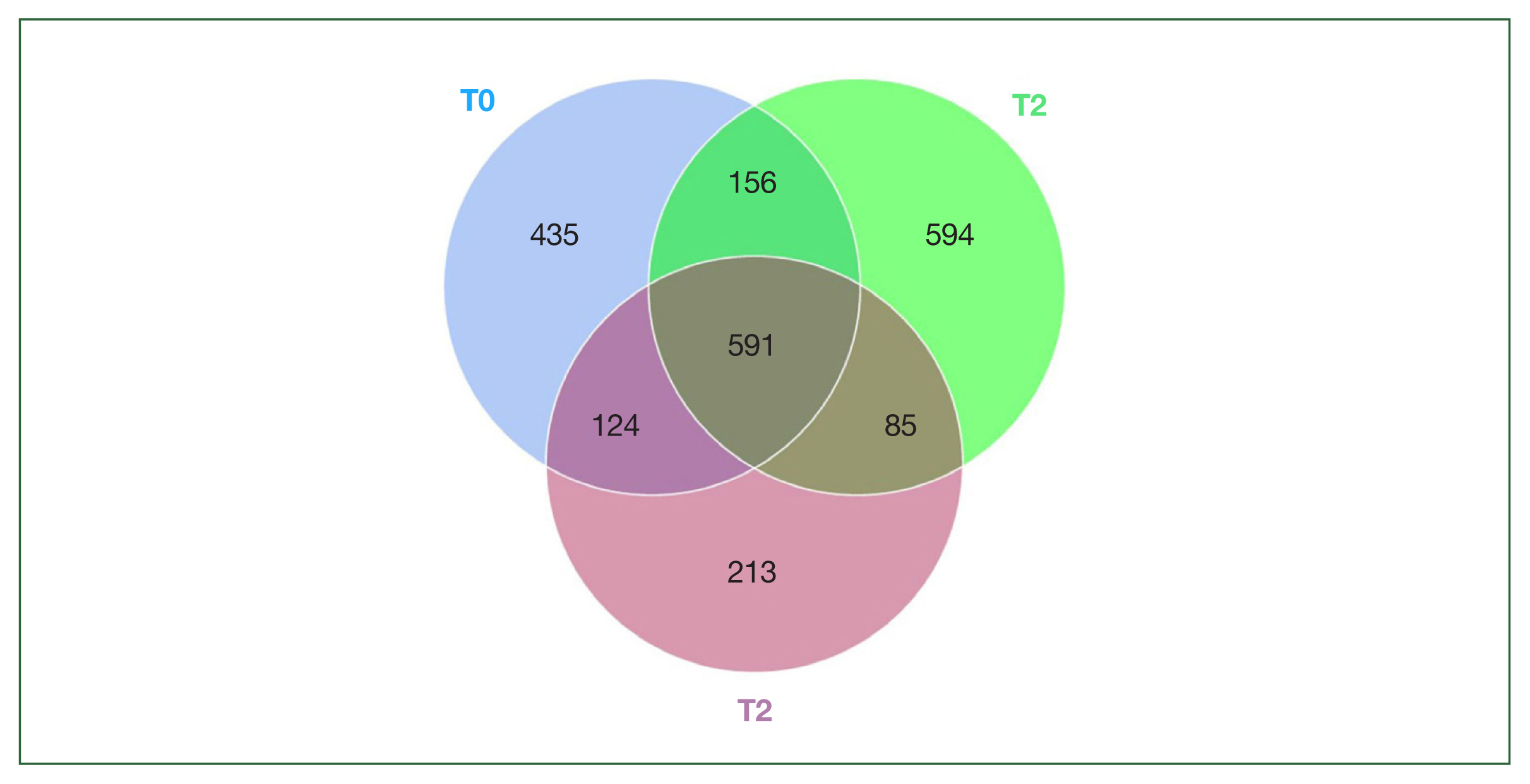
P-value=0.02), which number was reduced with the server of symptoms (T2 group, 1,013 ASVs). The composition of the bacterial community demonstrated distinctions among the 3 groups. In 3 groups, we found a total of 2,198 ASVs, of which 435,594, and 213 ASVs were unique in the T0, T1, and T2 groups, respectively. 892 ASVs were unique in
B. duncani infection groups. 591 ASVs were found in all groups (
Fig. 2).
Alpha rarefaction values demonstrated that all samples attained saturation, which indicated that species number was not rise with sample size (
Supplementary Fig. S1).
Fig. 3 depicts the bacterial community composition of the contents of small intensity in control and
B. duncani-infected hamsters. The stacked column diagram of species’ relative abundance was from the top 10 taxa, which made up>95% of the reads.
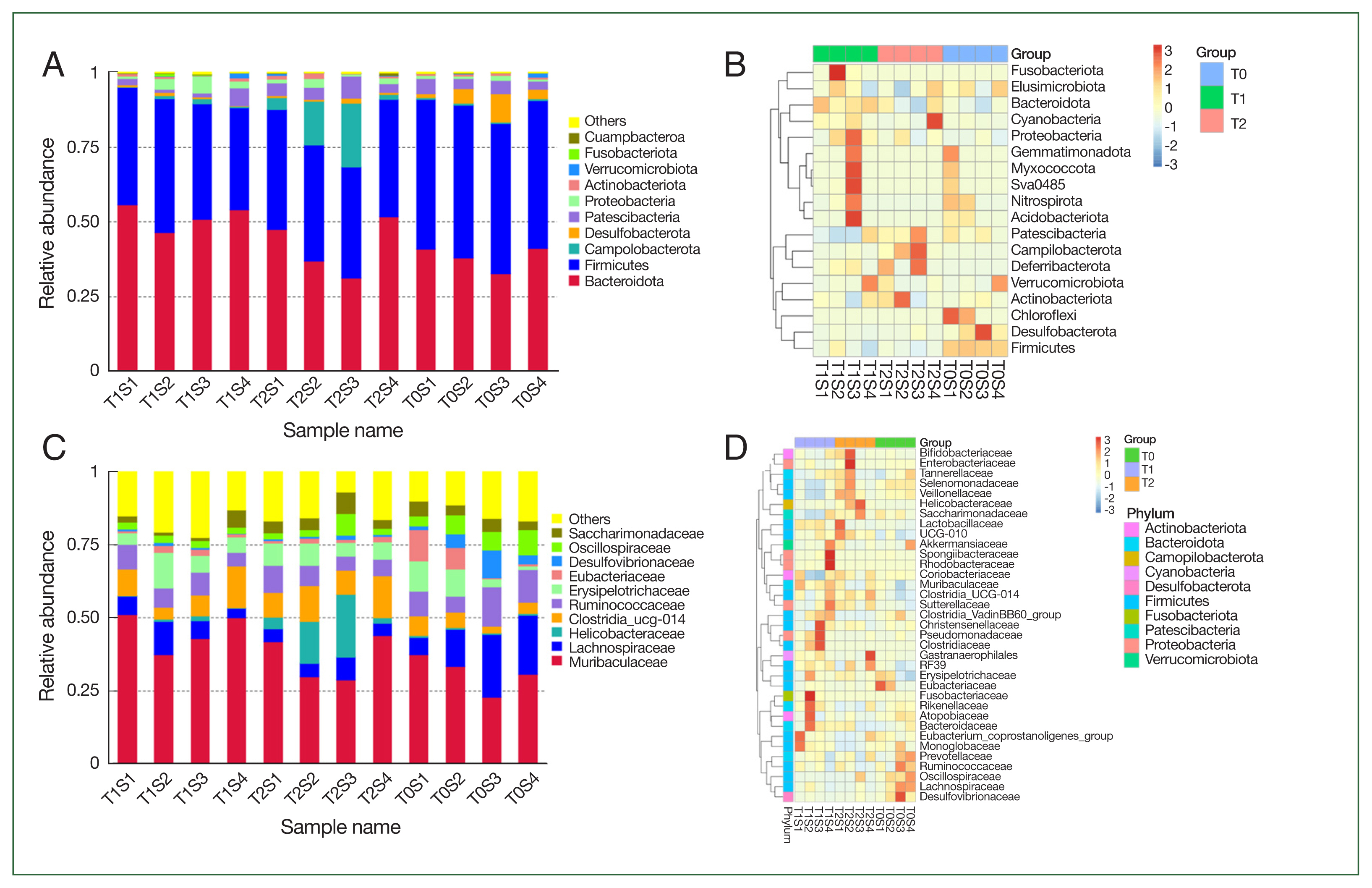
At the phylum, Bacteroidetes and Firmicutes comprised the main components of the gut microbiota in all groups. The relative abundance of Bacteroidetes rose from an average of 38% in the T0 group to 51.7% in the T1 group and 42% in the T2 group with the rise of the burden of parasite and the severity of disease, while the
Campilobacterota increased from 0.6% in T1 group to 0.8% in T1 and 1% in T2 groups. However,
Firmicutes and
Desulfobacterota showed a downtrend in T1 and T2 groups compared with T0 (
Fig. 3).
At the family level, the most common component in all samples was Muribaculaceae, which rose from 31% in the T0 group to 45% in T1 and 36% in the T2 groups. The relative abundance of Helicobacteraceae and Clostridia UCG014 also demonstrated similar phenome. While Lachnospiraceae, Eubacteriaceae, and Desulfovibrionaceae were considerably decreased after B. duncani infection (T2 group) compared with the T0 group with P-values of 0.038, 0.007, and 0.01.
Alpha diversity analyses
Alpha diversity was employed to assess the microbiota community in the samples. To evaluate the bacterial diversity, we used the Simpson diversity index of the alpha diversity parameter, implying the richness and relative abundance of ASVs. There is no significant difference among T0 (0.986±0.039), T1 (0.976±0.012), and T2 (0.989±0.045) groups (
P=0.53). The indexes of identified ASVs were similar between the T0 (650.75±30.73) and T1 (662.25±69.15) groups (
P=0.77), however significant variation was found in T2 (551.75±64.81) and T0/T1 groups (
P=0.03). Other indexes, including goods coverage, Chao1, Shannon, and phylogenetic diversity, were shown in
Table 2.
For weighted UniFrac and unweighted UniFrac determining changes in species, the beta-diversity was distinct between the T2 group and T0. The B. duncani infection group (T2) had a low number of ASVs than the uninfected group (T0) (P=0.02, t-test; P=0.015, Wilcoxon rank-sum test), implying that the beta-diversity increased from T0 to T2. However, for the weighted and unweighted UniFrac analysis about changes in species abundance, there was no substantial distinction between T1 and T0 (P=0.79, t-test; P=0.65, Wilcoxon rank-sum test). Overall, the beta-diversity reduced from T0 to T2.
According to weighted and unweighted UniFrac distance matrixes, the similarity of microbiota composition was examined using PCoA analyses. Microbiota changed significantly as the disease progressed in hamsters, induced by
B. duncani infection. PCoA analyses were conducted to identify the similarity degree between microbiota composition by evaluating within-group unweighted and weighted UniFrac distances. The microbiota of each group was distinct from other groups (
Fig. 4). Similar to the PCoA plot, distances within groups were substantially lower than between groups (ANOSIM, MRPP,
P=0.015). Irregularities among groups were examined using R-value (0.196), which demonstrated significant differences between groups. Findings from multi-response permutation procedure (MRPP) analysis revealed that a lower difference was found within the group and a higher difference was observed between groups. The value of MRPP was 0.046, implying that the differences among groups were higher than those within the group (A>0).
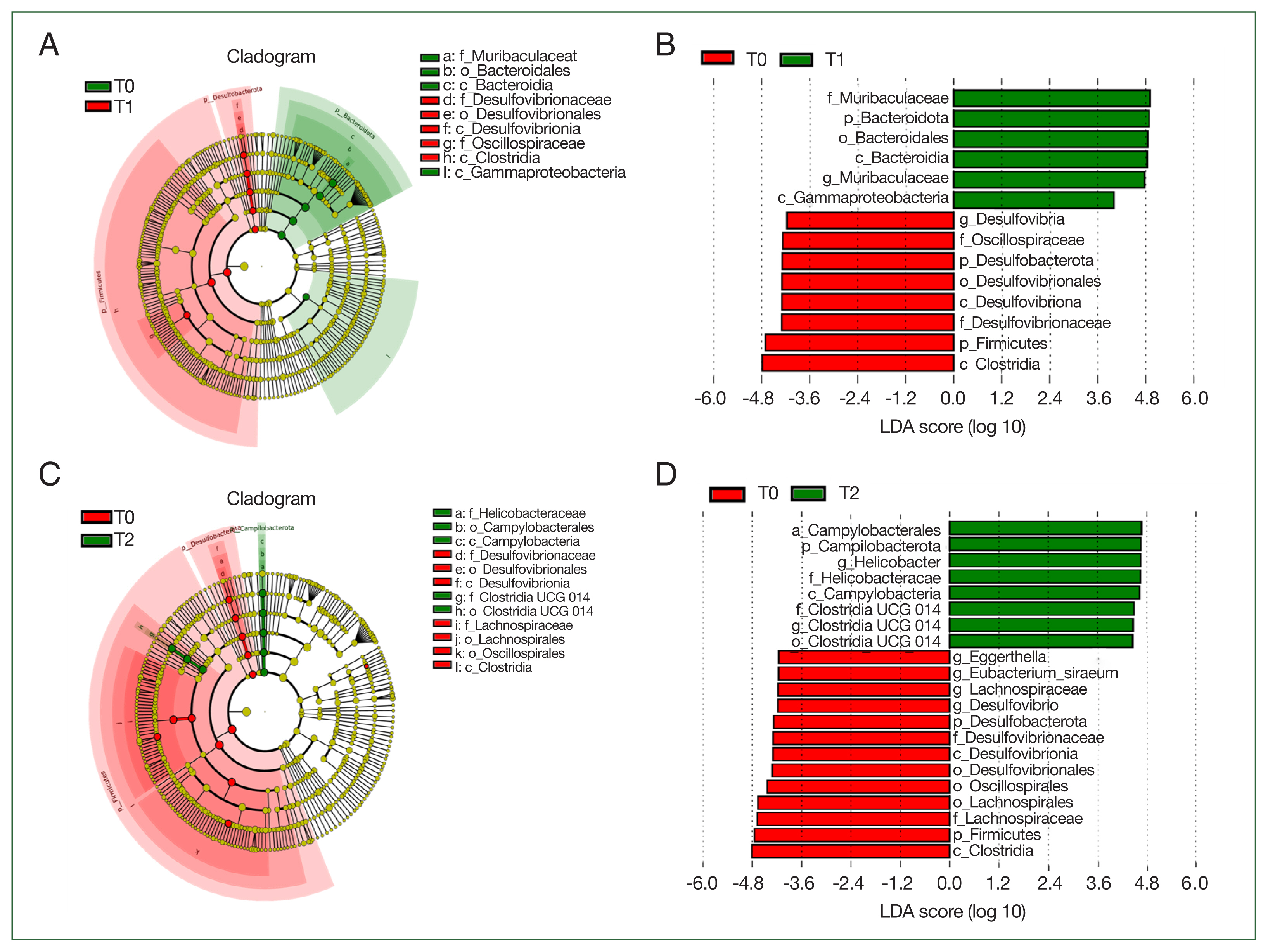
LEfSe analysis was employed to determine the biomarkers, which were bacterial taxa with substantial differences between groups (
Fig. 5). We conducted LEfSe analysis on the top 10 taxa. LDA score 4 was set as the criterion to evaluate the taxa in analyzed groups. The predicted biomarkers at different taxonomic levels were identified in T0, T1, and T2 groups. At the family level, 6 families (Muribaculaceae, Oscillospiraceae, Lactobacillaceae, Oscillospiraceae, Veillonellaceae, and Selenomonadaceae) were the notable difference between T0 and T1 groups; and 4 (Helicobacteraceae, Clostridia UGG014, Desulfovibrionaceae, and Lachnospiraceae) were identified between T0 and T2 groups.
Discussion
Babesiosis is a common infectious disease worldwide, particularly in tropical and subtropical regions. The clinical symptoms of
Babesia spp. infection range from subclinical illness to serve disease causing death, being linked to host immune status.
Babesia species co-infection with bacteria can cause fever, dark-colored urine, and respiratory distress syndrome [
15], however, limited research has been carried out to assess the effect of
Babesia spp. on the gut microbiota of hosts/experimental animals. Research has shown that the murine gut microbiome controls the severity of malaria in mice and effect on parasite burden of
Plasmodium rodent species [
10]. In mosquito vectors, a link between the gut microbial composition and
Plasmodium parasites has been found [
16–
18]. Research revealed that specific gut bacteria can against the transmission of
P. berghei from mosquitos to mice [
19]. In this research, we demonstrated that the
B. duncani infectious can affect the gut microbiome in hamsters, which was linked to the burden and severity of babesiosis.
Gut microbiota compositions in
B. duncani-infected and uninfected hamsters were analyzed. Sequencing data almost coverage of the total microbiota diversity due to species accumulation boxplots and rarefaction curves. According to the bacterial diversity analysis, we found that the diversity and relative abundance of the gut microbiota in all small intestine contents were modified after
B. duncani infection, while at the phylum level, Bacteroidetes and Firmicutes were dominated in all groups, which is similar to a previous study in
P. berghei infectious in mice [
12,
20]. Our data revealed that the relative abundance of the top 10 taxa altered after infection.
B. duncani infection increased the abundance of Bacteroidetes and Campilobacterota, than
B. duncani-free group T0. Our results differ from a previous study on
P. berghei infection in mice, in which researchers show an increase of Proteobacteria and Verrucomicrobia. Other research revealed that
P. berghei infection increased the Firmicutes and Proteobacteria in the feces [
21].
Simultaneously, the families Lachnospiraceae, Ruminococcaceae, Eubacteriaceae, Desulfovibrionaceae, and Oscillospiraceae were substantially decreased after
B. duncani infection than the T0 group. Lachnospiraceae may contribute to the production of short-chain fatty acids, which provide the energy source for intestinal epithelial cells. This family takes part in retaining the intestinal barrier and modulating gut motility [
21]. Increasing in this family is linked to metabolic syndrome in patients. Ruminococcaceae are also linked to metabolism and immune function [
22]. A lower abundance of Ruminococcaceae is found in acute-on-chronic liver failure. Eubacteriaceae, Desulfovibrionaceae, and Oscillospiraceae may partake in the breakdown of protein [
23,
24]. The underlying mechanism between
B. duncani infection and microbiota modifications should be further explored.
We conducted the LEfSe analysis to determine the biomarkers with great differences between
B. duncani-infected and
B. duncani-free groups. Our outcome provides a new understanding of host intestinal microbiota variation after
B. duncani infection. Nicolas et al. [
10] revealed that the abundance of Lactobacillus and Bifidobacterium was increased in resistant mice and probiotics, such as Lactobacillus and Bifidobacterium, influenced decreased parasite burden. Whether the biomarkers, found in this study, conduct a similar effect on pathogenesis is required to study. LEfSe analysis showed that the potential biomarkers of the T0 group were almost similar to T1 and T2 groups, while the potential biomarkers of the T2 group were greatly distinct from those of the T1 group. The possible reason was that potential biomarkers may have significant changes with the development of the disease.
To our best knowledge, this research is the first to evaluate the relationship between gut microbiota profile and B. duncani infection in hamsters and assess potential biomarkers involved in babesiosis. However, this research also has some limitations. We did not present underlying mechanisms linked to changes in gut bacterial composition, which should be investigated in the future. Conducting large-scale experimental animals in each group is proposed to present more efficient data. Whether modulation of microbiota profile can be an alternative strategy against B. duncani should be studied.
Notes
-
The authors declare no competing financial interests.
-
Author contributions
Data curation: Wang Y, Yueli N
Formal analysis: Zhang S
Funding acquisition: Guan G
Investigation: Wang Y
Methodology: Zhang S, Wang J, Li X
Supervision: You C, Zhang D, Guan G
Writing – original draft: Zhang S, Wang J
Supplementary Information
Supplementary Fig. S1
Bacterial community comparison of the groups. (A) alpha diversity analysis according to the Chao1 index; (B) alpha diversity analysis according to the observed amplicon sequence variant (ASV) index; (C) alpha diversity analysis according to the Shannon index; (D) alpha diversity analysis according to the Good’s coverage index.
Acknowledgments
This study was financially supported by the Science Fund for Creative Research Groups of Gansu Province (22JR5RA024), the Natural Science Foundation of Gansu (22JR5RA031), Gansu Natural Science Foundation (21JR11RA130), the Science and Technology Development Guiding Program of Lanzhou (2019ZD-55), Cuiying Scientific and Technological Program of Lanzhou University Second Hospital (CY2019-BJ05), the National Science Foundation of China (grant no. 31972701), ASTIP (grant no. CAAS-ASTIP-2016-LVRI), and NBCIS (grant no. CARS-37), the Leading Fund of Lanzhou Veterinary Research Institute (LVRI-SZJJ-202105), and the hatching program of SKLVEB (SKLVEB2021CGQD02).
Fig. 1Microscopy of Babesia duncani in hamsters. (A) the T0 group without parasite infection; (B) the T1 group with 2%±0.35% of parasitemia; (C) the T2 group with 20%±3.5% of parasitemia.

Fig. 2Microbial composition changes. Relative abundance of bacterial phyla and families in hamsters. Each bar suggests the top 10 most abundant phyla and families, while others imply unclassified and low-abundance ASVs. (A) bacterial community composition at the phylum; (B) taxonomic heatmap of the 3 groups at the phylum level; (C) bacterial community composition at the families; (D) taxonomic heatmap of the 3 groups at the families.
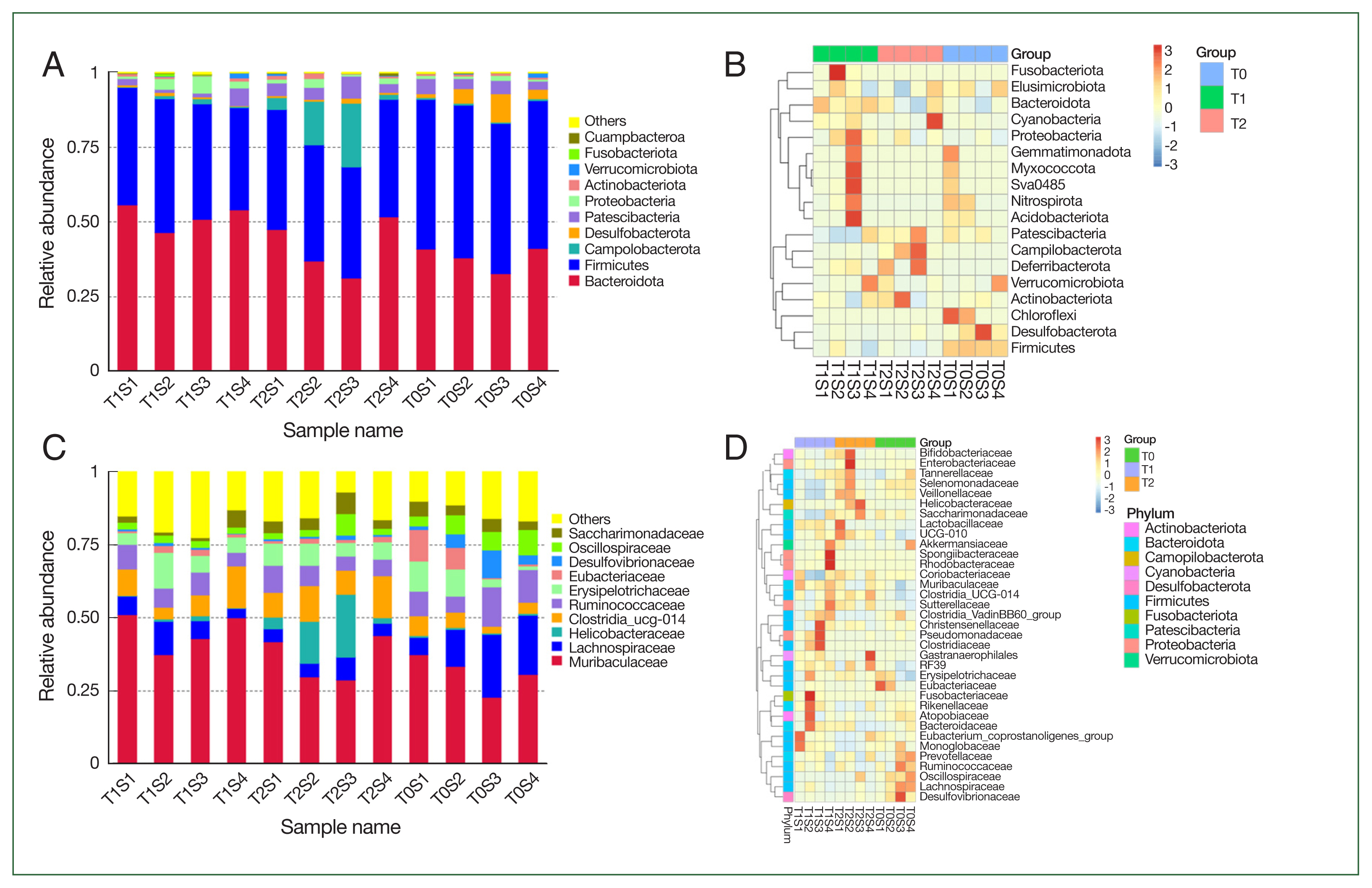
Fig. 3Operational taxonomic units (ASVs) analysis in the different samples.

Fig. 4Principal coordinates study based on weighted (A) and unweighted (B) UniFrac distance matrixes of microbiota profiles of hamsters infected by B. duncani.

Fig. 5Biomarkers in different groups. (A) cladogram of the most differentially abundant bacterial taxa in the 2 groups (T0 vs T1); (B), bacterial taxa that significantly differentiated in T0 vs T1 groups were determined by LEfSe analysis; (C), cladogram of the most differentially abundant bacterial taxa in the 2 groups (T0 vs T2); (D), bacterial taxa that significantly differentiated in T0 vs T2 groups were identified by LEfSe analysis.
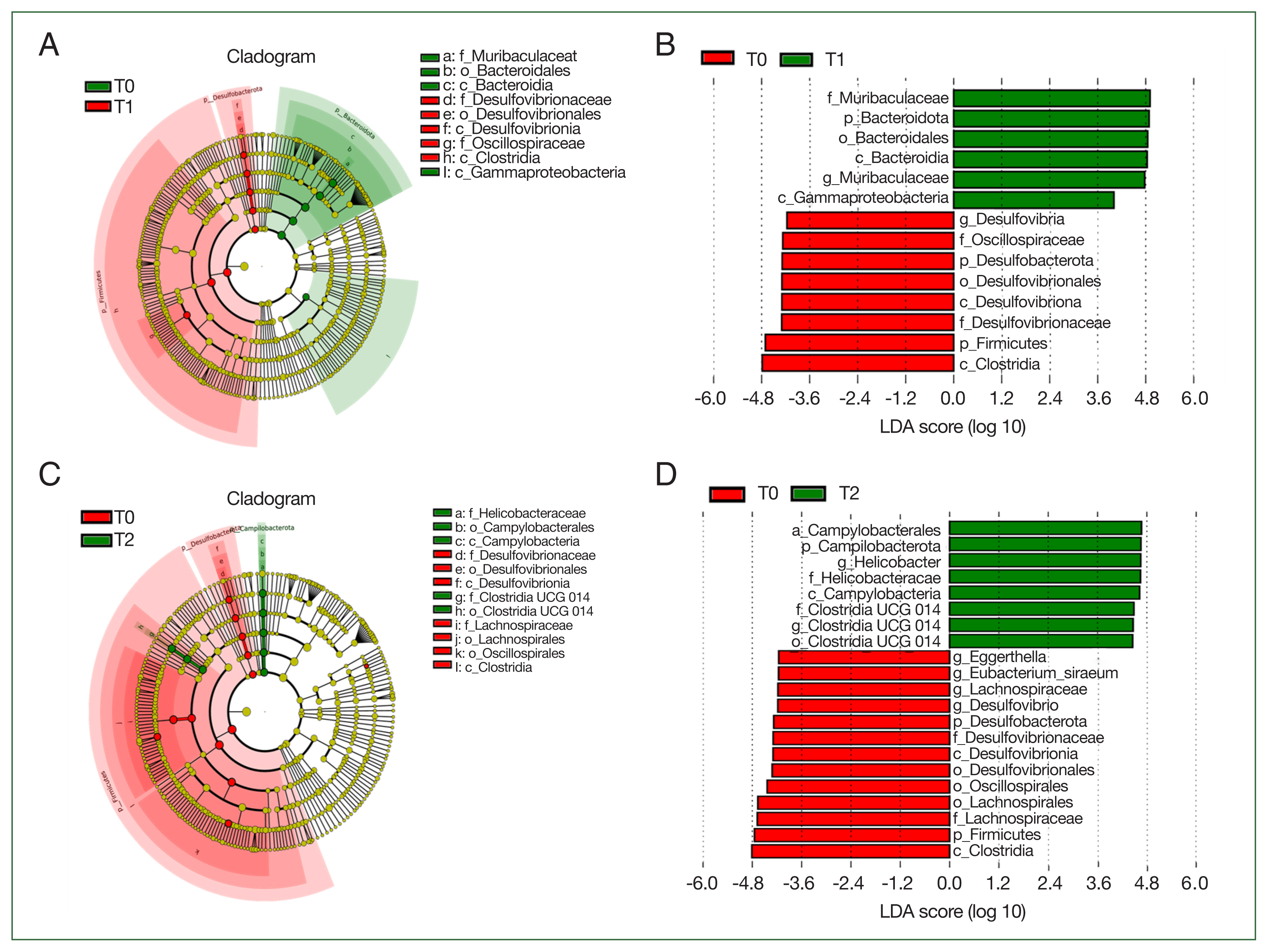
Table 1Basic information of the sequence data from infected and uninfected hamster small intestinal samples
Table 1
|
Group |
Sample name |
Qualified tags |
Nochime tags |
Average length (nt) |
Q20 |
Q30 |
GC (%) |
Effective tags (%) |
ASV |
|
T0 |
T0S1 |
101,060 |
68,583 |
417 |
96.92 |
91.17 |
54.2 |
66.7 |
608 |
|
T0S2 |
100,352 |
68,754 |
417 |
96.79 |
90.8 |
54.4 |
67.0 |
667 |
|
T0S3 |
107,321 |
70,960 |
415 |
97.01 |
91.3 |
54.2 |
64.9 |
650 |
|
T0S4 |
101,936 |
64,287 |
415 |
97.01 |
91.36 |
54.4 |
61.4 |
679 |
|
|
T1 |
T1S1 |
104,339 |
70,300 |
418 |
96.73 |
90.81 |
54.3 |
66.7 |
588 |
|
T1S2 |
108,420 |
71,697 |
419 |
96.56 |
89.93 |
54.1 |
64.9 |
740 |
|
T1S3 |
110,857 |
73,670 |
419 |
96.99 |
90.98 |
53.8 |
64.4 |
722 |
|
T1S4 |
107,882 |
78,769 |
418 |
96.83 |
91.05 |
54.2 |
72.1 |
599 |
|
|
T2 |
T2S1 |
90,804 |
61,839 |
418 |
96.67 |
90.55 |
53.9 |
66.1 |
445 |
|
T2S2 |
109,550 |
75,387 |
416 |
97.1 |
91.53 |
53.1 |
67.3 |
555 |
|
T2S3 |
104,938 |
76,067 |
413 |
97.17 |
91.69 |
52.9 |
71.3 |
608 |
|
T2S4 |
103,767 |
75,601 |
418 |
96.84 |
91.06 |
53.8 |
70.8 |
541 |
Table 2Alpha diversity indexes for the microbial communities detected in the hamster samples
Table 2
|
Group |
Sample name |
Chao1 |
Dominance |
Goods coverage |
Observed ASVs |
Pielou_e |
Shannon |
Simpson |
|
T0 |
T0S1 |
608 |
0.016 |
1 |
608 |
0.8 |
7.399 |
0.984 |
|
T0S2 |
668 |
0.013 |
1 |
667 |
0.814 |
7.638 |
0.987 |
|
T0S3 |
650.091 |
0.018 |
1 |
650 |
0.796 |
7.439 |
0.982 |
|
T0S4 |
678.125 |
0.009 |
1 |
678 |
0.843 |
7.928 |
0.991 |
|
|
T1 |
T1S1 |
588.214 |
0.01 |
1 |
588 |
0.826 |
7.601 |
0.99 |
|
T1S2 |
740.3 |
0.007 |
1 |
740 |
0.86 |
8.199 |
0.993 |
|
T1S3 |
722.25 |
0.008 |
1 |
722 |
0.842 |
7.996 |
0.992 |
|
T1S4 |
599.077 |
0.017 |
1 |
599 |
0.786 |
7.252 |
0.983 |
|
|
T2 |
T2S1 |
445 |
0.018 |
1 |
445 |
0.803 |
7.064 |
0.982 |
|
T2S2 |
555.273 |
0.026 |
1 |
555 |
0.776 |
7.078 |
0.974 |
|
T2S3 |
609.667 |
0.04 |
1 |
608 |
0.747 |
6.906 |
0.96 |
|
T2S4 |
541.333 |
0.012 |
1 |
541 |
0.819 |
7.434 |
0.988 |
References
- 1. Gray J, Zintl A, Hildebrandt A, Hunfeld KP, Weiss L. Zoonotic babesiosis: overview of the disease and novel aspects of pathogen identity. Ticks Tick Borne Dis 2010;1(1):3-10. https://doi.org/10.1016/j.ttbdis.2009.11.003
- 2. Cornillot E, Hadj-Kaddour K, Dassouli A, Noel B, Ranwez V, et al. Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti. Nucleic Acids Res 2012;40(18):9102-9114. https://doi.org/10.1093/nar/gks700
- 3. Skrabalo Z, Deanovic Z. Piroplasmosis in man; report of a case. Doc Med Geogr Trop 1957;9(1):11-16.
- 4. Scholtens RG, Braff EH, Healey GA, Gleason Gleason N. A case of babesiosis in man in the United States. Am J Trop Med Hyg 1968;17(6):810-813. https://doi.org/10.4269/ajtmh.1968.17.810
- 5. Wang J, Zhang S, Yang J, Liu J, Zhang D, et al. Babesia divergens in human in Gansu province, China. Emerg Microbes Infect 2019;8(1):959-961. https://doi.org/10.1080/22221751.2019.1635431
- 6. Kim JY, Cho SH, Joo HN, Tsuji M, Cho SR, et al. First case of human babesiosis in Korea: detection and characterization of a novel type of Babesia sp. (KO1) similar to ovine Babesia. J Clin Microbiol 2007;45(6):2084-2087. https://doi.org/10.1128/JCM.01334-06
- 7. Shih CM, Liu LP, Chung WC, Ong SJ, Wang CC. Human babesiosis in Taiwan: asymptomatic infection with a Babesia microti-like organism in a Taiwanese woman. J Clin Microbiol 1997;35(2):450-454. https://doi.org/10.1128/jcm.35.2.450-454.1997
- 8. Quick RE, Herwaldt BL, Thomford JW, Garnett ME, Eberhard ML, et al. Babesiosis in Washington State: a new species of Babesia? Ann Intern Med 1993;119(4):284-290. https://doi.org/10.7326/0003-4819-119-4-199308150-00006
- 9. Scott JD, Scott CM. Human babesiosis caused by Babesia duncani has widespread distribution across Canada. Healthcare (Basel) 2018;6(2):49. https://doi.org/10.3390/healthcare6020049
- 10. Villarino NF, LeCleir GR, Denny JE, Dearth SP, Harding CL, et al. Composition of the gut microbiota modulates the severity of malaria. Proc Natl Acad Sci U S A 2016;113(8):2235-2240. https://doi.org/10.1073/pnas.1504887113
- 11. Kamada N, Chen GY, Inohara N, Núñez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 2013;14(7):685-690. https://doi.org/10.1038/ni.2608
- 12. Fan ZG, Li X, Fu HY, Zhou LM, Gong FL, et al. Gut microbiota reconstruction following host infection with blood-stage Plasmodium berghei ANKA strain in a murine model. Curr Med Sci 2019;39(6):883-889. https://doi.org/10.1007/s11596-019-2119-y
- 13. Wei N, Cao J, Zhang H, Zhou Y, Zhou J. The tick microbiota dysbiosis promote tick-borne pathogen transstadial transmission in a Babesia microti-infected mouse model. Front Cell Infect Microbiol 2021;11:713466. https://doi.org/10.3389/fcimb.2021.713466
- 14. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 2019;37(8):852-857. https://doi.org/10.1038/s41587-019-0209-9
- 15. Abrams Y. Complications of coinfection with Babesia and Lyme disease after splenectomy. J Am Board Fam Med 2008;21(1):75-77. https://doi.org/10.3122/jabfm.2008.01.060182
- 16. Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, et al. Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 2011;332(6031):855-858. https://doi.org/10.1126/science.1201618
- 17. Dong Y, Manfredini F, Dimopoulos G. Implication of the mosquito midgut microbiota in the defense against malaria parasites. PLoS Pathog 2009;5(5):e1000423. https://doi.org/10.1371/journal.ppat.1000423
- 18. Azambuja P, Garcia ES, Ratcliffe NA. Gut microbiota and parasite transmission by insect vectors. Trends Parasitol 2005;21(12):568-572. https://doi.org/10.1016/j.pt.2005.09.011
- 19. Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell 2014;159(6):1277-1289. https://doi.org/10.1016/j.cell.2014.10.053
- 20. Taniguchi T, Miyauchi E, Nakamura S, Hirai M, Suzue K, et al. Plasmodium berghei ANKA causes intestinal malaria associated with dysbiosis. Sci Rep 2015;5:15699. https://doi.org/10.1038/srep15699
- 21. Guan W, Yang S, Zhao Y, Cheng W, Song X, et al. Observation of the gut microbiota profile in C57BL/6 mice induced by Plasmodium berghei ANKA infection. Front Cell Infect Microbiol 2021;11:680383. https://doi.org/10.3389/fcimb.2021.680383
- 22. Johnson EL, Heaver SL, Walters WA, Ley RE. Microbiome and metabolic disease: revisiting the bacterial phylum Bacteroidetes. J Mol Med (Berl) 2017;95(1):1-8. https://doi.org/10.1007/s00109-016-1492-2
- 23. Raimondi S, Musmeci E, Candeliere F, Amaretti A, Rossi M. Identification of mucin degraders of the human gut microbiota. Sci Rep 2021;11(1):11094. https://doi.org/10.1038/s41598-021-90553-4
- 24. Shimizu Y, Nakamura K, Kikuchi M, Ukawa S, Nakamura K, et al. Lower human defensin 5 in elderly people compared to middle-aged is associated with differences in the intestinal microbiota composition: the DOSANCO health study. Geroscience 2022;44(2):997-1009. https://doi.org/10.1007/s11357-021-00398-y

 , Jinming Wang2,†
, Jinming Wang2,†