Abstract
Ticks host different pathogens as endosymbiont and nonpathogenic microorganisms and play an important role in reproductive fitness and nutrient provision. However, the bacterial microbiomes of white-tailed deer ticks have received minimal attention. This study aimed to examine the bacterial microbiome of ticks collected from Odocoileus virginianus on the Mexico–United States border to assess differences in microbiome diversity in ticks of different species, sexes, and localities. Five different tick species were collected: Rhipicephalus microplus, Dermacentor nitens, Otobius megnini, Amblyomma cajennense, and A. maculatum. The tick microbiomes were analyzed using next-generation sequencing. Among all tick species, the most predominant phylum was Proteobacteria, followed by Actinobacteria and Firmicutes. The ticks from Tamaulipas and Nuevo León presented the highest bacterial species diversity. Acinetobacter johnsonii and A. lwoffii were the common bacterial species in the microbiome of all ticks, Coxiella were present in R. microplus, and Dermacentor nitens also exhibited a Francisella-like endosymbiont. The microbiome of most females in D. nitens was less diverse than that of males, whereas R. microplus occurs in females, suggesting that microbiome diversity is influenced by sex. In the bacterial communities of A. maculatum and O. megnini, Candidatus Midichloria massiliensis, and Candidatus Endoecteinascidia fumentensis were the most predominant endosymbionts. These results constitute the initial report on these bacteria, and this is also the first study to characterize the microbiome of O. megnini.
-
Key words: Odocoileus virginianus, Rhipicephalus, Dermacentor, Otobius, Amblyomma, tick, white-tailed deer, endosymbionts, microbiome
Introduction
Ticks are obligate hematophagous ectoparasites of vertebrates, and blood of various hosts, including humans and animals, is their sole source of nutrition. Ticks harbor different pathogens such as
Borrelia,
Coxiella,
Francisella, and
Rickettsia, endosymbionts such as
Coxiella-like and
Francisella-like endosymbionts (CLEs and FLEs, respectively), and nonpathogenic microorganisms. These microorganisms may have a major role as efficient vectors and could also have an important function in reproductive fitness and nutrient provision [
1,
2]. The importance of microbial communities in ticks derives from their association with increased or decreased colonization and transmission of virulent pathogens [
3]. This transmission occurs either directly through nutrient competition and induced/reduced immunity or indirectly by affecting tick viability, reproduction, and physical condition. Studies on the relationships between ticks, their vertebrate hosts, and their microbial community (including endosymbionts and pathogens) have generally received minimal attention; indeed, the role of tick endosymbionts in pathogen transmission has been studied in only a few bacterial and tick species [
1]. However, additional research is necessary to understand the functions of nonpathogenic microorganisms as their intracellular biological cycle makes them difficult to isolate [
1,
2].
Microbial diversity in ticks is influenced by factors such as the environment (laboratory or wild), species, instar, feeding state, collection season, geographic region, and presence of pathogens [
1]. In addition, several microorganisms have a transovarial transmission route, where the maternal microbiota can serve as the first inoculum for developing eggs and larvae. Copulation and the microbiota on the host skin may also contribute to this diversity [
4].
White-tailed deer (
Odocoileus virginianus) play a dual role in the survival and proliferation of ticks, serving as a preferred food source and as a transporting vehicle for ticks to locate their preferred habitat [
5]. The intensive management of wild or semicaptive white-tailed deer populations, which often share space with cattle in ranches, enhances hunting income in game farms in northern Mexico but also increases zoonotic pathogen risks [
6]. The main legal white-tailed deer–hunting farms are located on the border between the United States and Mexico, through the Mexican states of Sonora, Chihuahua, Coahuila, Nuevo León, and Tamaulipas, and deer usually occupy the same pasture areas as cattle [
7]. Therefore, infectious diseases can be shared since bacterial communities can be transmitted from deer to cattle (or vice versa) and occasionally to humans [
8].
Herein, a high-throughput sequencing approach was used to examine the bacterial microbiome of the digestive tract of ticks collected from O. virginianus in northern locations of Mexico to assess the differences in microbiome diversity and abundance in ticks of different sexes, species, and study localities.
Materials and methods
Ethics statement
No ethical approval was required. All samples were collected from legally hunted animals during the hunting season.
Tick collection and identification
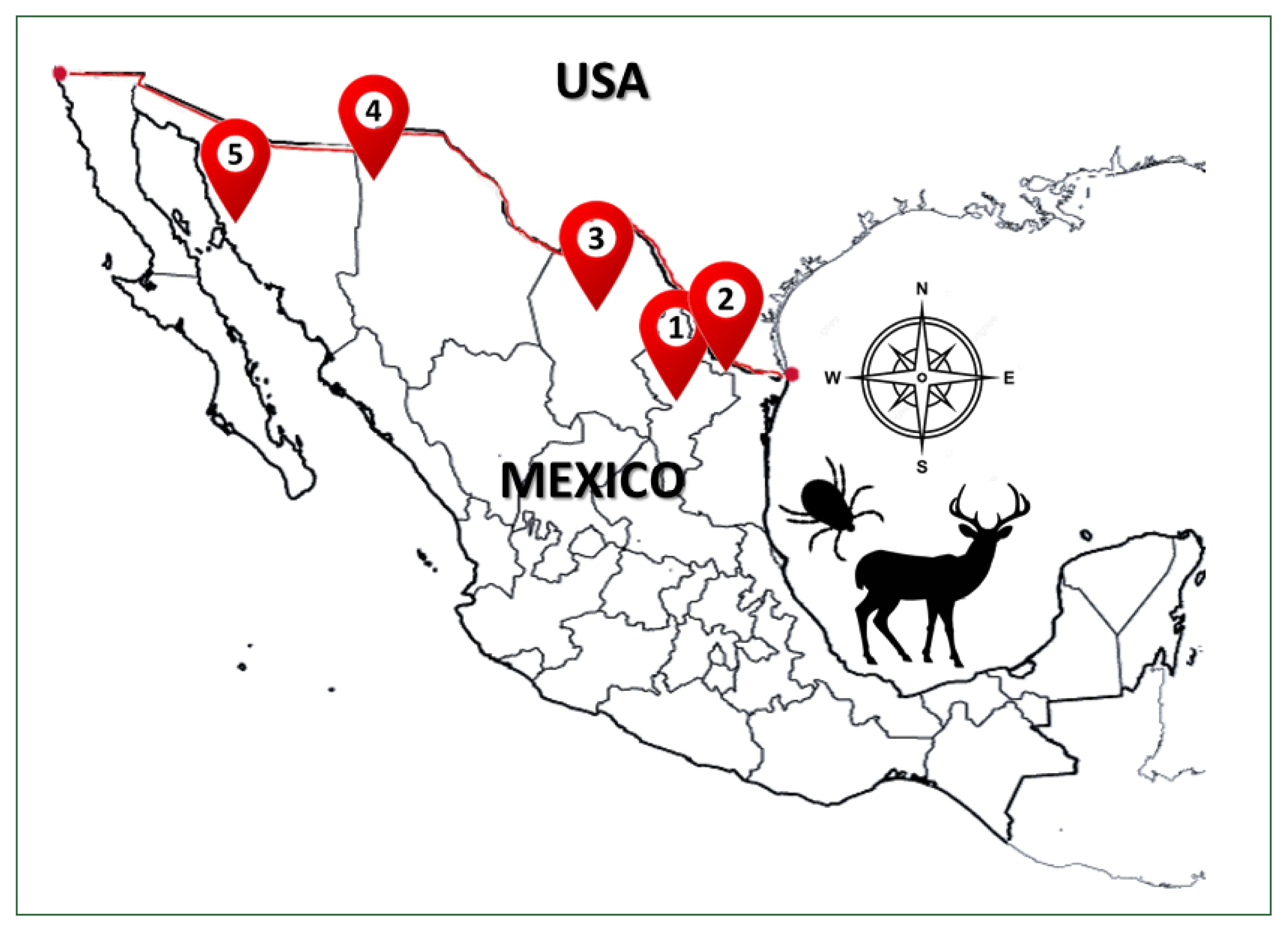
Ticks were collected from 57 of 296 white-tailed deer during the legal hunting season (November 2018–February 2020) in 5 northern states of Mexico—Sonora (Son), Chihuahua (Chih), Coahuila (Coah), Nuevo León (NL), and Tamaulipas (Tam)—by authorization from the Ministry of Environment and Natural Resources of Mexico (
Fig. 1). The geographical coordinates of each ranch and municipality are described in
Table 1 [
9]. All tick collections were made in collaboration with teams from Organización de Vida Silvestre [Wildlife Organization], whose staff accompanied hunters. Sterilized tweezers were used to remove ticks from the most populated area on captured hosts in a 5-min period. Specimens were kept alive until dissection or preserved in jars with 96% ethanol and processed in the Molecular and Experimental Pathology Laboratory of the Biological Sciences Faculty at the Autonomous University of Nuevo León. Taxonomic identification was performed by morphology under a 10× to 40× stereoscope (EZ4E, Leica Microsystem, Jalisco, Mexico) using pictorial keys [
10]. In total, 14 males and 126 females of
Rhipicephalus microplus, 74 females and 49 males of
Dermacentor nitens, 6 males of
Amblyomma cajennense, 2 males of
A. maculatum, and 19 nymphs of
Otobius megnini were obtained. One vial per tick species containing 2–10 digestive tracts was randomly selected for microbiome analyses according to sex and locality.
The external surface of the ticks was sterilized in 3% H
2O
2, rinsed with 70% ethanol, and subsequently rinsed three times in sterile saline solution for 1 min [
11]. To collect the digestive tract samples, the abdominal cuticle was sliced aseptically using sterilized forceps and a disposable scalpel blade (size #11) under a stereoscope in a biohazard cabinet to avoid cross-contamination. DNAzol (Molecular Research Center, Cincinnati, OH, USA) was used for DNA extraction with the protocol previously modified in our laboratory [
12], and then quantified with a Nanodrop 2000 (Thermo Fisher Scientific Inc., Waltham, MA, USA) and stored at −20°C. Molecular-grade water was used as a negative extraction control, and DNA extraction steps were performed under the same conditions.
Production of the 16S rDNA library, sequencing, raw read curation, and processing were achieved using the ZymoBIOMICS service (Zymo Research, Irvine, CA, USA). In brief, DNA samples were prepared for targeted sequencing using the Quick-16S NGS Library Prep Kit (Zymo Research). Bacterial 16S primers amplified the V3–V4 region of the 16S rRNA gene [
13] in a quantitative real-time PCR (qPCR) thermocycler to control cycles and, therefore, limit PCR chimera formation. Positive and negative controls for qPCR quality assessment were included during amplification. Final PCR products were fluorescence quantified and pooled on the basis of equal molarity. The pooled library was cleaned using the Select-a-Size DNA Clean & Concentrator Kit (Zymo Research) and then quantified using TapeStation (Agilent Technologies, Santa Clara, CA, USA) and Qubit (Thermo Fisher Scientific, Waltham, WA, USA). The final library was sequenced using Illumina MiSeq (Illumina, San Diego, CA, USA) with 600 cycles of a v3 sequencing kit (2×250-bp paired-end reads). Sequencing was performed with >10% PhiX spike-in.
Unique or distinctive amplicons were inferred from the raw reads and grouped into operational taxonomic units (OTUs) using the DADA2 pipeline [
14] after chimeric sequences were removed. Taxonomy assignment was performed with UCLUST from QIIME v1.9.1. using the Zymo Research. Database as a reference and the Greengenes 16S database Microbial community diversity (Shannon diversity) was determined using QIIME v1.9.1 [
15], and Student’s
t-test was performed to compare diversity among species, localities, and sexes. SIMPER analysis was performed to determine the main families that contributed to the dissimilarity in microbiome diversity among tick species. Principal coordinate analyses (PCoAs) were performed on the basis of Bray–Curtis distances and visualized using three-dimensional plots in EMPeror (a part of QIIME). Taxonomies with significant abundance among different groups were identified via linear discriminant analysis effect size (LEfSe) using default settings [
16].
A Microbial Community DNA Standard (Zymo Research) and blank library preparation samples were included as positive and negative controls, respectively.
Results
A total of 296 deer were inspected at the studied locations, 57 (19.3%) of which were infested with ticks. In total, 290 ticks were collected and identified into 5 different species or complexes:
R. microplus,
D. nitens,
O. megnini,
A. cajennense, and
A. maculatum. Significantly more (
P<0.05) female ticks (200; 69%) were found than male ticks (71; 24.4%). Female ticks belonged only to
R. microplus (from NL, Tam, Coah, and Chih) and
D. nitens (from NL, Coah, and Chih). All other ticks were males of
A. cajennense and
A. maculatum (6 and 2, respectively), and 19 (6.5%) were nymphal stadia of
O. megnini (
Table 1).
The metagenomic dataset of this study provides information on the microbiome diversity of the 5 collected tick species and a comparative analysis between sexes and among localities. A total of 1,035,712 raw read sequences (R1+R2) were processed from the 14 samples to obtain 537 unique OTUs, which were identified as observed species after trimming, removal of chimeric sequences, and size filtration. Female
R. microplus (Tam F) with full microbiome profiles exhibited higher bacterial richness (number of observed species), with 86 final unique sequences, followed by male
A. cajennense (NL M), with 69 identified sequences (
Supplementary Fig. S1A), showing significant differences among them (Student,s
t-test
P<0.007). When comparing between sexes per locality, a significantly higher richness was only found in
R. microplus females (NL F) than that in males (42 and 11 OTUs, respectively; Student,s
t-test
P<0.001) (
Supplementary Fig. S1B) (samples from Coah, Tam, and Chih included only females and thus were excluded). Conversely, male
D. nitens from the three different localities where this was found (Chih, NL, and Coah with 57, 32, and 19 OTUs, respectively) showed a higher bacterial richness than that in females (18, 31, and 18 OTUS, respectively) with significant differences due to specimens from Chih (57 OTUs) (Student,s
t-test
P<0.0001) (
Table 2;
Supplementary Fig. S1C).
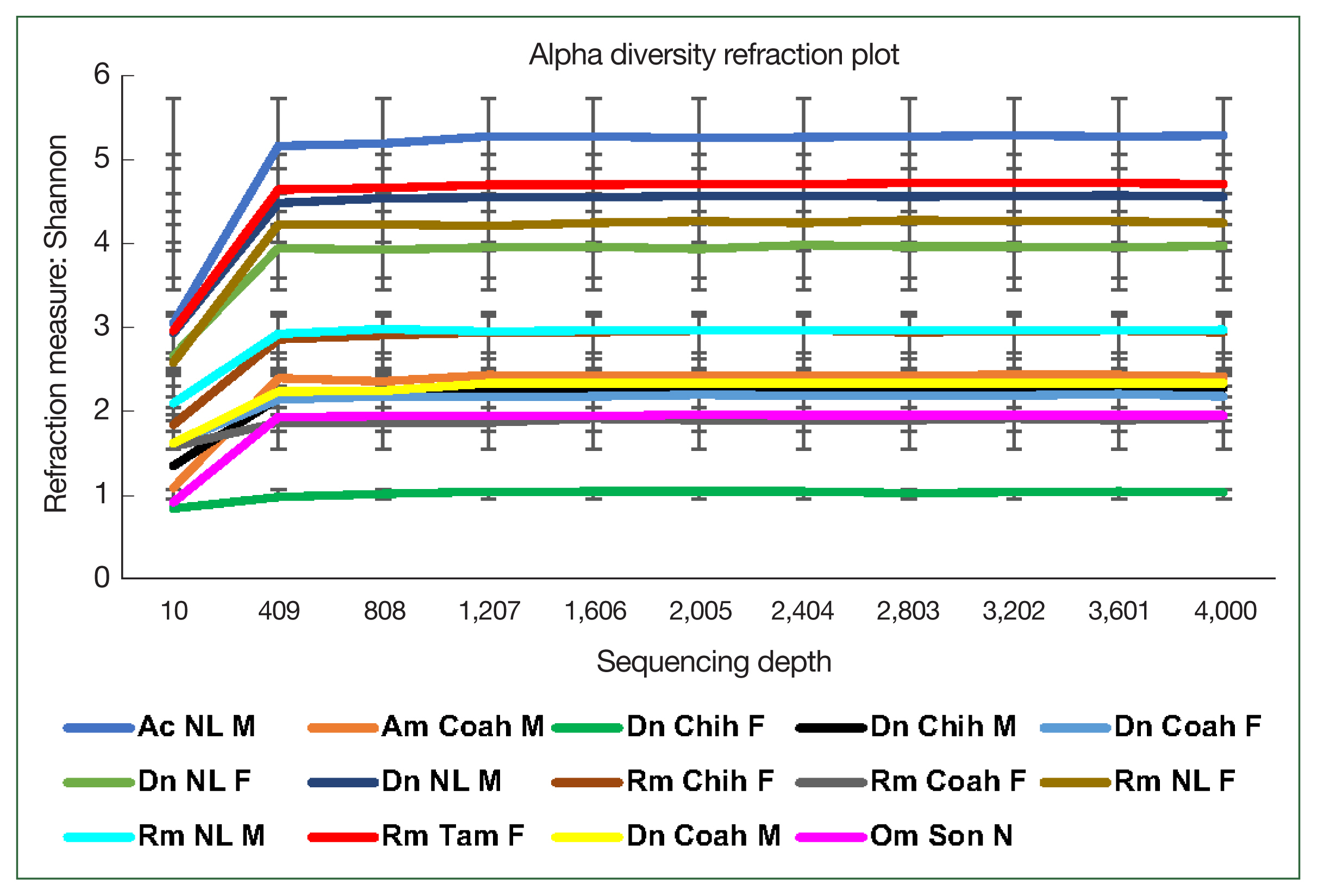
The rarefaction curve represents the alpha bacterial diversity (Shannon index,
Fig. 2) and bacterial richness at a depth of 4,000 sequences per sample. These results suggest sufficient sequencing depth, as shown by plateauing of each sample, demonstrating that the sequencing captured most of the abundant bacterial species and that further sequencing would not detect new haplotypes or bacterial species. Regarding the comparative diversity of the rarefaction measure among the tick species, the bacterial diversity (Shannon index) in
A. cajennense (5.26) was notably higher than that in the other samples, and the lowest diversity was observed in
O. megnini (1.9). The comparison of alpha diversity between sexes showed similar results with those obtained with other measures of bacterial richness, where
D. nitens males (NL M) showed higher indexes than female ticks of all species, with the exception of a
R. microplus female (NL F) (
Fig. 2). Concerning the comparative analysis by locality,
R. microplus (Tam) had the highest diversity (5.26), followed by
D. nitens (NL) and
R. microplus (NL) (4.75 and 4.55, respectively) (
Fig. 2).
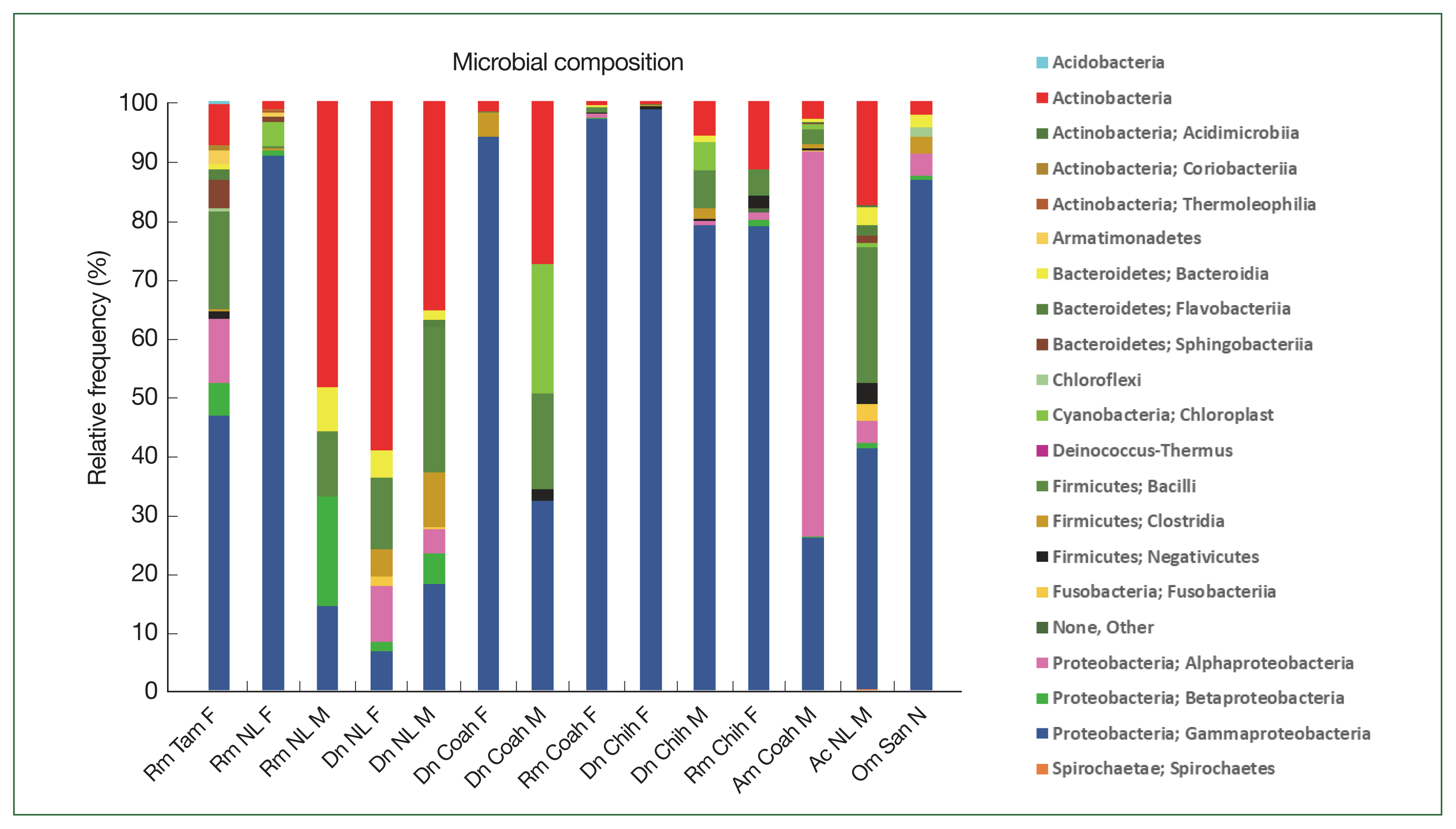
The class Gammaproteobacteria (phylum Proteobacteria) had the highest abundance in almost all tick groups analyzed by locality and sex at 98.6%, 96.9%, 94%, 90.4%, and 86.8% in D. nitens (Chih F), R. microplus (Coah F), D. nitens (Coah F), R. microplus (NL F), and O. megnini, respectively, followed by Alphaproteobacteria (10.8% in R. microplus, Tam F, and 3.7% in O. megnini), and Betaproteobacteria (R. microplus, Tam F, with 5.5%). However, the class Alphaproteobacteria had the highest abundance in A. maculatum (65.5%), followed by Actinobacteria in D. nitens NL F and NL M (59.2% and 35.5%, respectively).
In
R. microplus (NL M), the abundance of Proteobacteria reached 48.5%, whereas the abundance of Firmicutes reached 24.6% in
D. nitens (NL M) and 22.9% in
A. cajennense (NL M). The other minor phyla found in
A. cajennense were Fusobacteria (2.8%) and Bacteroidetes (1.3%); more detailed information is provided in
Fig. 3. The main bacterial orders in most of the species were Thiotrichales (Gammaproteobacteria, 86.8% and 98.6% in
O. megnini and
D. nitens, respectively); Rickettsiales (65.4% in
A. maculatum); and Pseudomonadales (33.8% and 26.5% in
A. cajennense and
R. microplus, respectively) (
Supplementary Table S1).
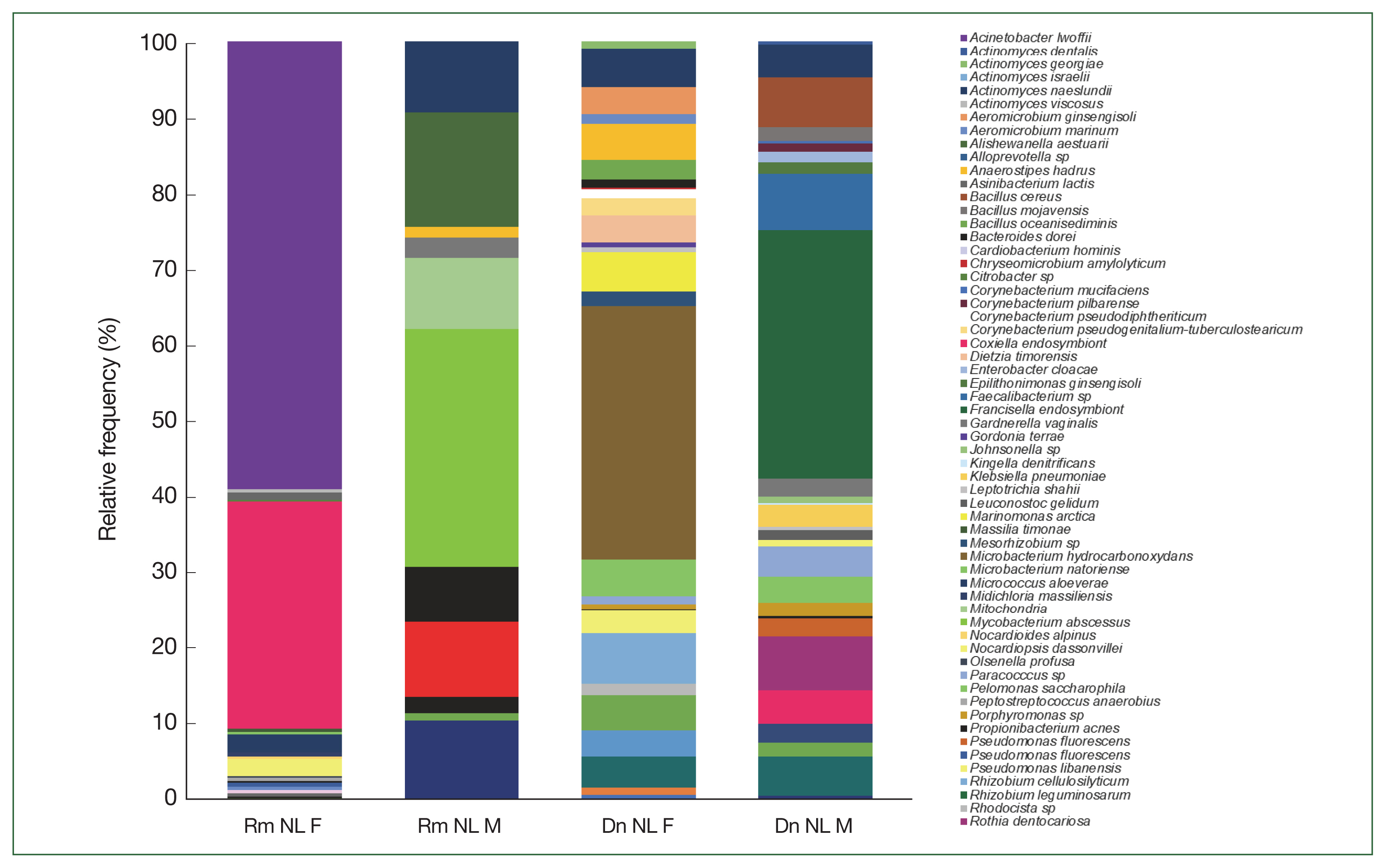
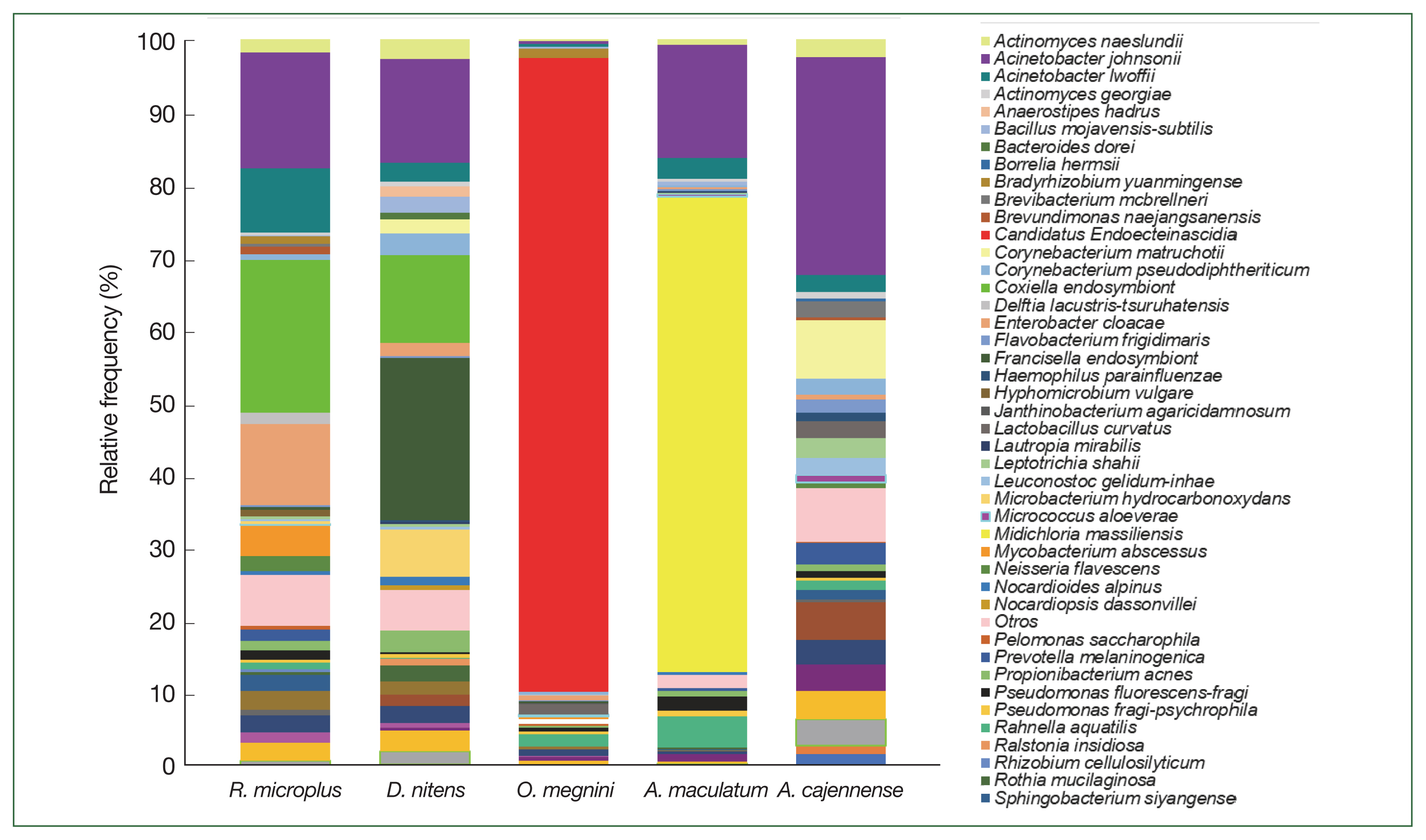
The relative frequency of the most abundant bacterial species identified could only be compared between females and males of
R. microplus and
D. nitens from NL (
Fig. 4) since only male ticks of
A. cajennense and
A maculatum were collected while only nymphs were found in
O. megnini (
Table 1). In male
R. microplus,
Mycobacterium abscessus, and
Alishewanella estuari were present at the highest relative abundance (30.0% and 14.4%, respectively), and in female
R. microplus,
Acinetobacter lwoffii had the highest relative abundance (58.6%), followed by CLE (29.8%).
Microbacterium hydrocarbonoxydans,
Rhizobium cellulosilyticum, and
Acinetobacter johnsonii were the most predominant species (32.2%, 6.5%, and 3%, respectively) in female
D. nitens among the 31 OTUs found in their microbiome. The bacterial richness of male
D. nitens was higher than that of females (32 OTUs,
Table 2), where the most abundant species were FLE,
Faecalibacterium sp., and
Bacillus cereus (32.2%, 7.4%, and 6.4%, respectively (
Supplementary Table S2).
At the genus level of tick microbiomes,
A. johnsonii (30%) accounted for almost a third of the composition of the
A. cajennense microbiome, being the most predominant, with
Borrelia hermsii (0.4%) and
Corynebacterium matruchotii (8%). CLE (21%),
A. johnsonii (15.9%),
Enterobacter cloacae (11.2%), and
A. lwoffii (8.9%) had the highest relative abundance in
R. microplus, and FLE,
A. johnsonii, CLE, and
M. hydrocarbonoxydans constituted the top 4 species in
D. nitens.
Candidatus Endoecteinascidia frumentensis (87.2%) was the dominant species in the microbiome of
O. megnini. Furthermore,
Midichloria massiliensis had the highest relative abundance (65.4%) in
A. maculatum (
Fig. 5). SIMPER analyses showed the dissimilarity values between tick species and identified the microbiome bacterial species that contributed to the differences (
Table 2). The highest significant dissimilarities (>80) were between
O. megnini and
A maculatum,
D. nitens,
A. cajennense, and
R. microplus (
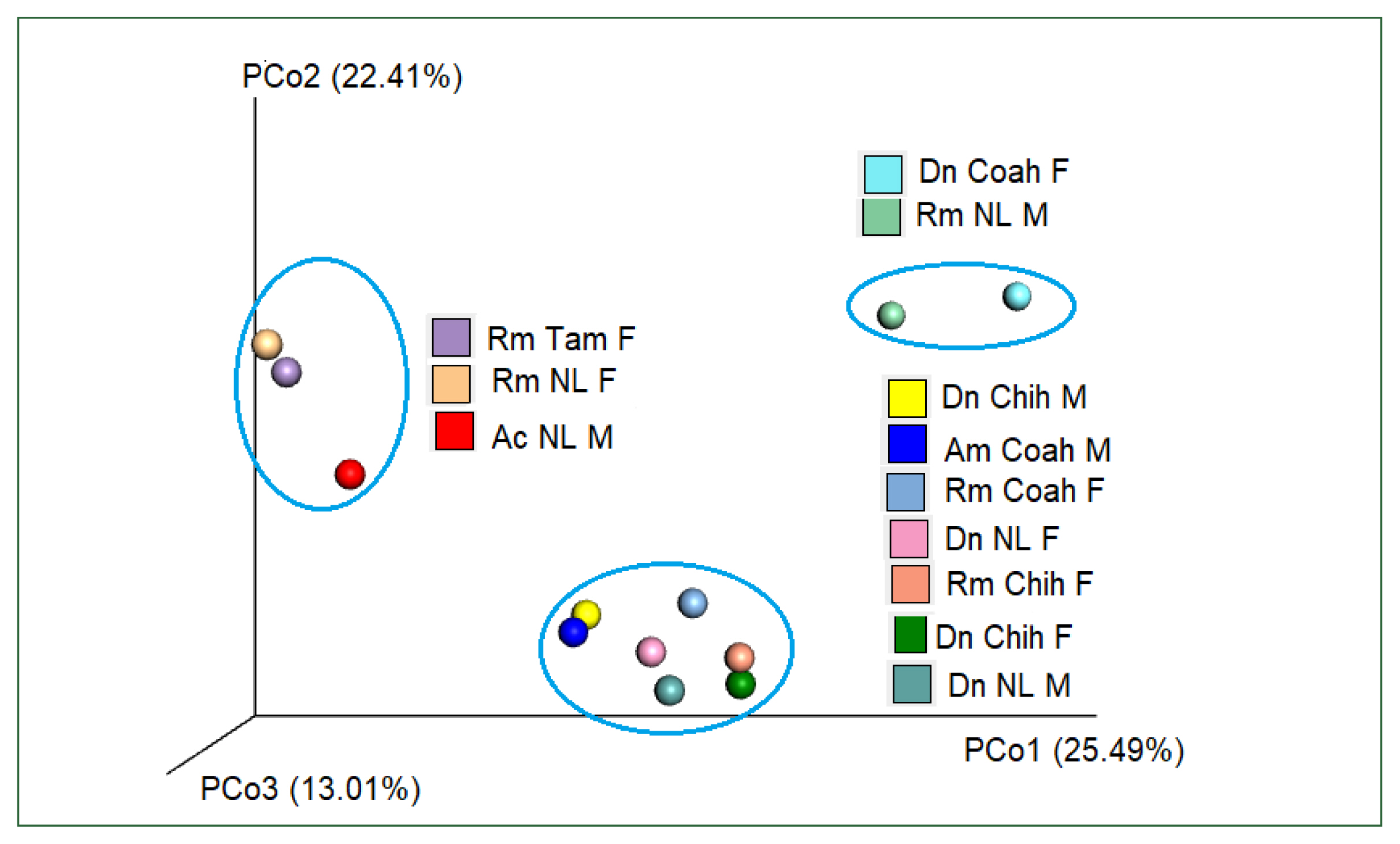
Table 3). PcoA using Bray–Curtis dissimilarity (beta diversity) of the bacterial community revealed that the first 2 axes (Pco1 and Pco2) explained 22.41% and 25.49% of the variation in the data, respectively. Thus, the samples of Dn Chih M, Dn Chih F, Dn NL M, Dn NL F, Rm Coah F, Rm Chih F, and Am Coah M (rank of similarity 72–86%) clustered separately from those of Rm NL F, Rm Tam F, and Ac NL M, demonstrating that the bacterial composition of the 2 groups differed (
Fig. 6). Raw sequence data are available in NCBI GenBank under Bioproject PRJNA88132 under accession numbers SRX18276467, SRX18276466, SRX17608462, SRX17608463, SRX18276468, SRX18276469, SRX17608464, SRX17608465, SRX17608466, SRX17608467, SRX17608468, SRX17608469, and SRX18275422.
Discussion
This study reports the microbial diversity of 5 tick species (
R. microplus,
D. nitens,
A. cajennense,
A. maculatum, and
O. megnini), ectoparasites of white-tailed deer from northern Mexico.
O. virginianus plays a crucial role as an alternative host for vectors that may carry many human and animal pathogens of zoonotic importance, including numerous endosymbionts and commensals [
17]. The density of ticks can be highly variable and depends on the season when they are collected and the geographical location and age of the ungulate hosts [
18]. Herein, 4 tick species were found as adults, except
O. megnini, which is only parasitic in its nymphal stadium [
19]. Three species of ticks were previously reported in
O. virginianus in the United States:
A. maculatum,
R. microplus, and
D. nitens [
20] while
O. megnini and
A. cajennense are described here for the first time from northern Mexico.
The bacterial microbiome of ticks from white-tailed deer was dominated by the phylum Proteobacteria, followed by Actinobacteria, Firmicutes, and Bacteroidetes, similar to the microbiome reported in
R. microplus from livestock in Antioquia [
21] since
R. microplus is the tick species most commonly found infesting cervius,
O. v. yucatanensis [
22]. No publications have described the bacterial diversity of
D. nitens,
A. cajennense,
A. maculatum, or
O. megnini specifically infesting
O. virginianus, which indicates that this is the first study on this topic.
The dominant eubacterial families identified included
Moraxellaceae with the genus
Acinetobacter, which has been characterized as a soil and environmental microorganism reported in tick species, such as
Ixodes scapularis,
Haemaphysalis flava,
A. americanum, and
D. niveus [
23].
Acinetobacter johnsonii was observed as a common bacterial species in the microbiome of all 5 tick species analyzed and was most frequently observed in
R. microplus (Coah F, 85.8%), followed by
D. nitens (Coah F, 60.7%),
A. cajennense (30%),
R. microplus (Chih F, 25.4%),
A. maculatum (15.6%), and
D. nitens (Chih 1.2–4.3%). In addition to
A. johnsonii,
A. leoffii was also highly prevalent and was found in
R. microplus, in the
Amblyomma complex (15.6–30%), in
D. nitens (1.2–4.3%), and at the 0.5% baseline in
O. megnini. The presence of
Acinetobacter in ticks is considered to indicate microbiome dysbiosis because of increased colonization of this microorganism, suggesting that
Acinetobacter may have an important role in the microbiome in decreasing tick susceptibility to environmental pollutants [
24]. Members of the families
Coxiellaceae and
Francisellaceae with endosymbionts of the genera CLE and FLE were observed in
R. microplus and
D. nitens as previously described [
25] in wild ticks from India, horse ticks, and livestock ticks from China [
26]. Functional studies have shown that CLE contributes to tick biology by affecting reproductive fitness and molting [
27]. Many
Coxiella species have been recorded as endosymbionts in ticks; for example, in
Amblyomma, their presence in the salivary glands alters the transmission of
Ehrlichia chaffeensis [
28].
Coxiella burnetii is considered an important pathogen of vertebrates and could be an exception within the genus, although the pathogenic potential remains under investigation [
29]. The bacterial microbiome of
D. nitens contained a member of the family
Francisellaceae [
25]. An association between FLE and
Dermacentor ticks has been demonstrated, and FLE survive because of transovarial transmission, although both FLE and CLE could have an ancestral origin from an infected vertebrate host [
25,
26].
Our results revealed that the order Rickettsiales was the most predominant in the microbiome of
A. maculatum, with many endosymbionts and reported the presence of
M. massiliensis (65.4%) for the first time;
M. massiliensis is considered a nonpathogenic species and has been identified in
I. ricinus [
30]. The bacterial community of
O. megnini mainly comprised the endosymbiont
Candidatus E. frumentensis (87.2%) in the nymphal stadium, which had not been previously reported in ticks; indeed, this is the first study to characterize the microbiome of
O. megnini and to report the presence of
Candidatus E. frumentensis.
Midichloria massiliensis showed the highest relative abundance (65%) in the microbiome of
A. maculatum. The high abundance of endosymbionts such as
Candidatus Midichloriaceae mitochondrii, which has been documented to limit next-generation sequencing efficacy in arthropods by masking less abundant bacteria, including pathogens [
31], could explain the low rarefaction index in our results.
Herein, greater bacterial richness and diversity (Shannon index) were observed in most male ticks (
D. nitens) than in female ticks, whereas the opposite was observed in
R. microplus, suggesting that the microbiome diversity is affected by sex. In
R. microplus,
A. lwoffii (58.6%) and CLE (29.8%) were observed only in females (NL), whereas
M. abscessus (30%) and
A. aestuarii (14.4%) were found in males. In
D. nitens,
M. hydrocarbonoxydans (32.2%) and
R. cellulosilyticum (6.5%) had the highest relative frequency in females (NL) since FLE and
Faecalibacterium sp. were the most predominant species in males (32.2% and 7.4%, respectively;
Supplementary Table S2). These results agree with those of a report on male
I. scapularis where males showed a higher microbiome diversity than females [
32]. However, in ticks, the composition of endosymbionts can fluctuate according to the geographic region, morphological state, and feeding period when they are collected as described for female
R. microplus from
Pecari tajacu in Peru, which showed greater bacterial richness and diversity [
33].
The greatest contribution to the differences between
O. megnini and the other tick groups were the bacterial species belonging to Thiotrichales, Midichloriaceae, Moraxellaceae, and
Francisellaceae. The genera FLE and CLE had a higher relative load with a major contribution to SIMPER, which may be explained by a role similar to that reported for
Rickettsia [
34] since these bacteria are vertically inherited via transovarial transmission [
35]. Their efficiency may be related to the degree of infection within the tick since “mass-infected” ticks universally produce infected offspring and “mild infections” that cause variable rates of transovarial transmission [
36], which is an advantageous adaptation for microorganisms that use this type of transmission [
34].
A comparison of the microbiomes of
R. microplus and
D. nitens among localities identified differences in microbial community composition that could be due to geographic region and environment [
1]. However, of the various environmental microbiota that ticks can access, only a few become “bona fide” microbiome members [
4].
In conclusion, we report the microbiome of 5 tick species (R. microplus, D. nitens, A. cajennense, A. maculatum, and O. megnini) infesting O. virginianus. Female ticks of R. microplus (Tam F) with full microbiome profiles exhibited the highest bacterial richness (86 OTUs) followed by that of males of A. cajennense from NL (69 OTUs), whereas males of R. microplus (NL) exhibited only 11 OTUs. The bacterial diversity (Shannon index) in A. cajennense (5.26) was notably higher than that in the other samples, and the lowest diversity was observed in O. megnini (1.9). The class Gammaproteobacteria had the highest abundance in almost all tick groups analyzed by locality and sex; however, the class Alphaproteobacteria had the highest abundance in A. maculatum. At the species level, A. johnsonii and A. lwoffii were the most dominant bacterial species in the microbiome of all analyzed ticks. In the bacterial communities of A. maculatum and O. megnini, Candidatus Midichloria massiliensis, and Candidatus E. fumentensis were the most predominant endosymbionts. This is also the first study to characterize the microbiome of O. megnini.
Notes
-
The authors have no competing interests to be declared.
-
Conceptualization: Molina-Garza ZJ
Formal analysis: Molina-Garza ZJ, Galaviz-Silva L
Funding acquisition: Galaviz-Silva L
Investigation: Cuesy-León M, Rosales-Encina JL
Methodology: Cuesy-León M, Baylón-Pacheco L
Project administration: Galaviz-Silva L
Supervision: Molina-Garza ZJ, Galaviz-Silva L
Validation: Molina-Garza ZJ, Baylón-Pacheco L
Visualization: Baylón-Pacheco L
Writing – original draft: Molina-Garza ZJ, Galaviz-Silva L
Writing – review & editing: Rosales-Encina JL, Galaviz-Silva L
Supplementary Information
Supplementary Fig. S1.
Student’s t test between localities and sexes of white-tailed deer ticks. (A) Bacterial richness (number of OTUs) between
Rhipicephalus microplus (Tam F) and A.
cajennense (NL M). (B) Comparison between male and female ticks of Rm NL, where females showed a significant difference. (C) Male ticks of Dermacentor nitens, in contrast, showed higher richness than females (GraphPad Software, version 9.5.1).
https://www.graphpad.com/quickcalcs/contMenu/). Statistical significance with Student’s t test is cited. (For abbreviations, see
Fig. 2).
Supplementary Table S2.
Relative frequency of the microbiome composition of females and males ticks Rhipicephalus microplus and Dermacentor nitens (NL).
Acknowledgments
We thank the staff from Organización de Vida Silvestre S.A. de C.V. (OVIS) for their participation during tick collection. This research was supported by FOINS PN-CONACyT (grant #3157) and PAICyT-UANL. We are also grateful to CONACyT for the scholarship granted to the second author (MCL) during her Ph.D. degree. Raw sequence data are available in NCBI GenBank under Bioproject PRJNA88132.
Fig. 1Geographic distribution of sampled ticks collected from white-tailed deer Odocoileus virginianus from 5 states north of Mexico country: 1) Nuevo Leon, 2) Tamaulipas, 3) Coahuila, 4) Chihuahua, and 5) Sonora.
Fig. 2Alpha diversity of the microbiome of female (F) and male (M) ticks Rhipicephalus microplus (Rm), Dermacentor nitens (Dn), Amblyomma cajennense (Ac), A. maculatum (Am), and nymphs (N) of Otobius megnini (Om) from Chiuahua (Chih), Coahuila (Coah), Nuevo Leon (NL), and Tamaulipas (Tam). The refraction curve shows the Shannon index with the coverage of OTUs. Bars indicate standard deviations (n=10, only in Am n=2).
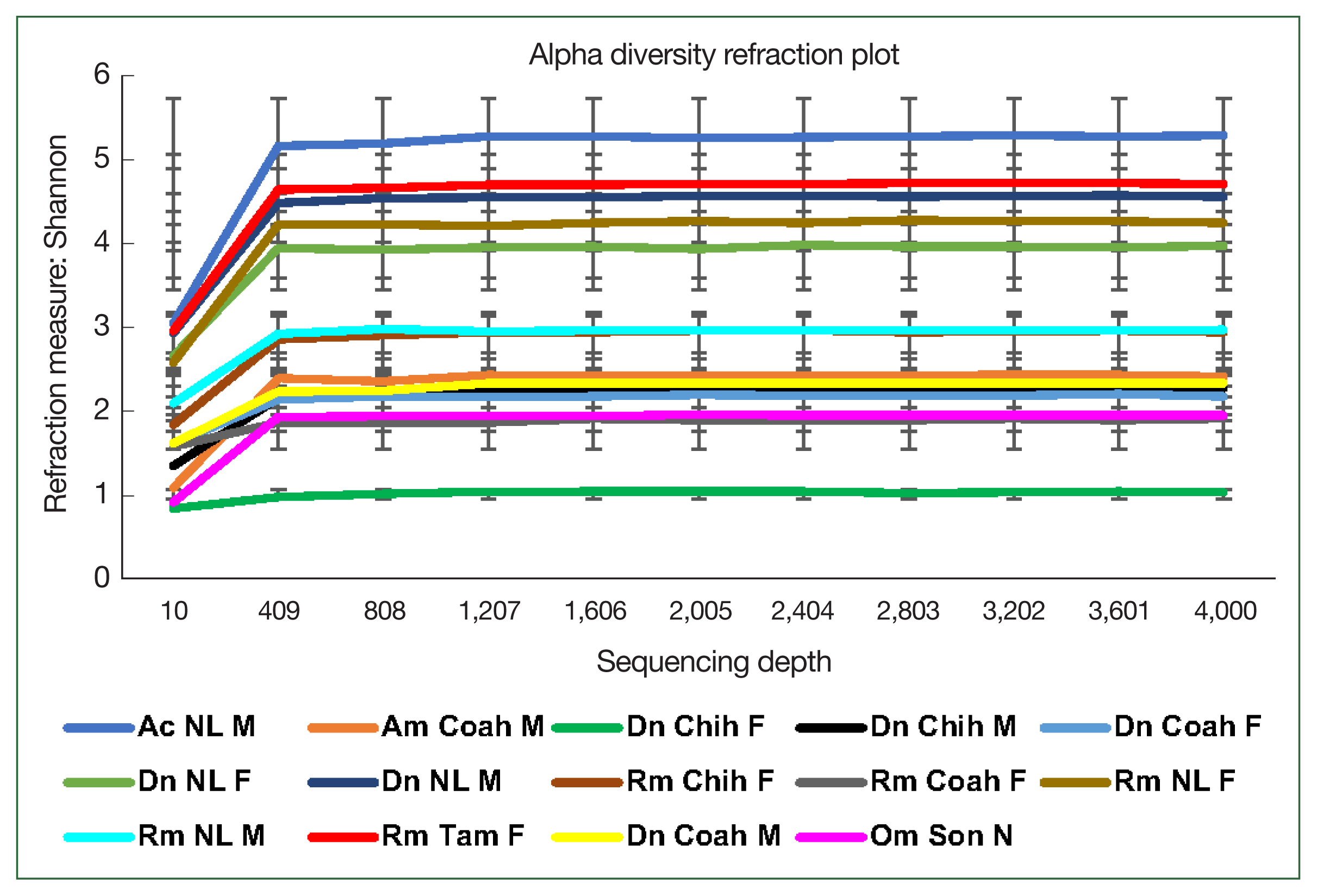
Fig. 3Relative frequency of the microbiome composition in the ticks at phylum level by gender and locality of
Rhipicephalus microplus,
Dermacentor nitens,
Amblyomma cajennense,
A. maculatum, and
Otobius megnini. For abbreviations, see
Fig. 2.
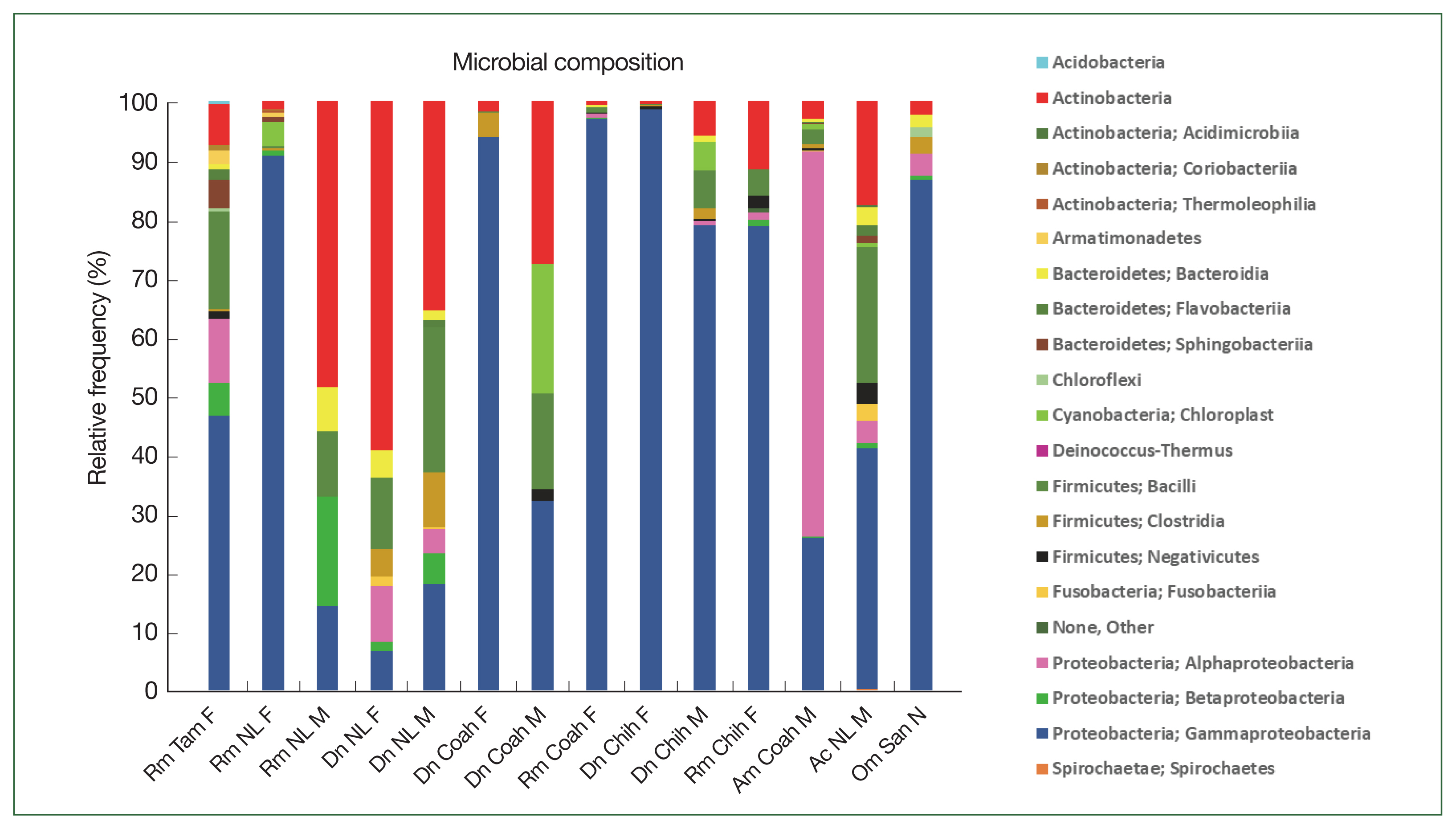
Fig. 4Relative frequency of the microbiome composition of ticks compared among female and male ticks of
Rhipicephalus microplus and
Dermacentor nitens (NL) at the species level with the most abundant species identified in the ticks. For abbreviations, see
Fig. 2.
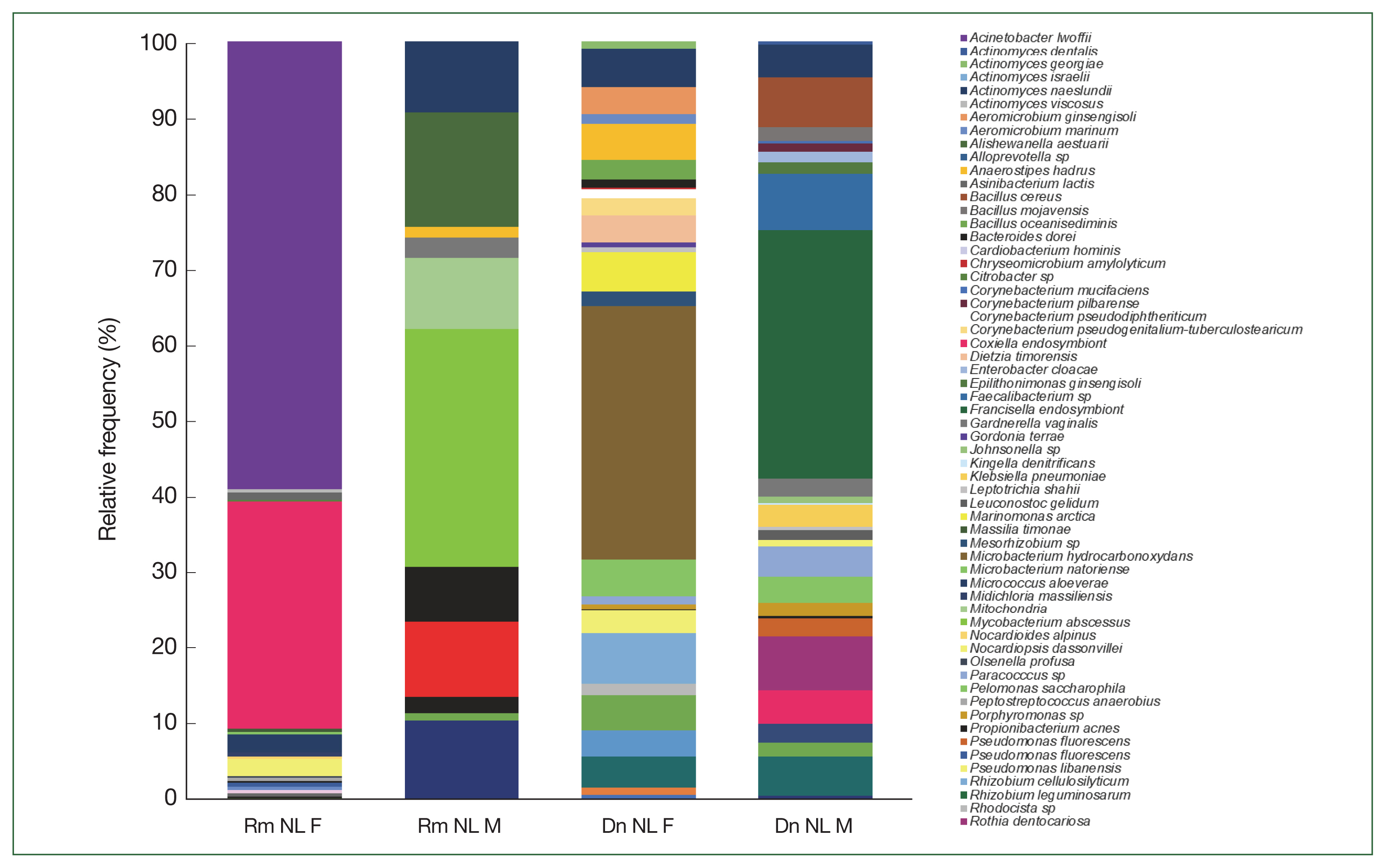
Fig. 5The relative frequency of the most abundant bacterial species identified in the 5 ticks collected from the white-tailed deer.
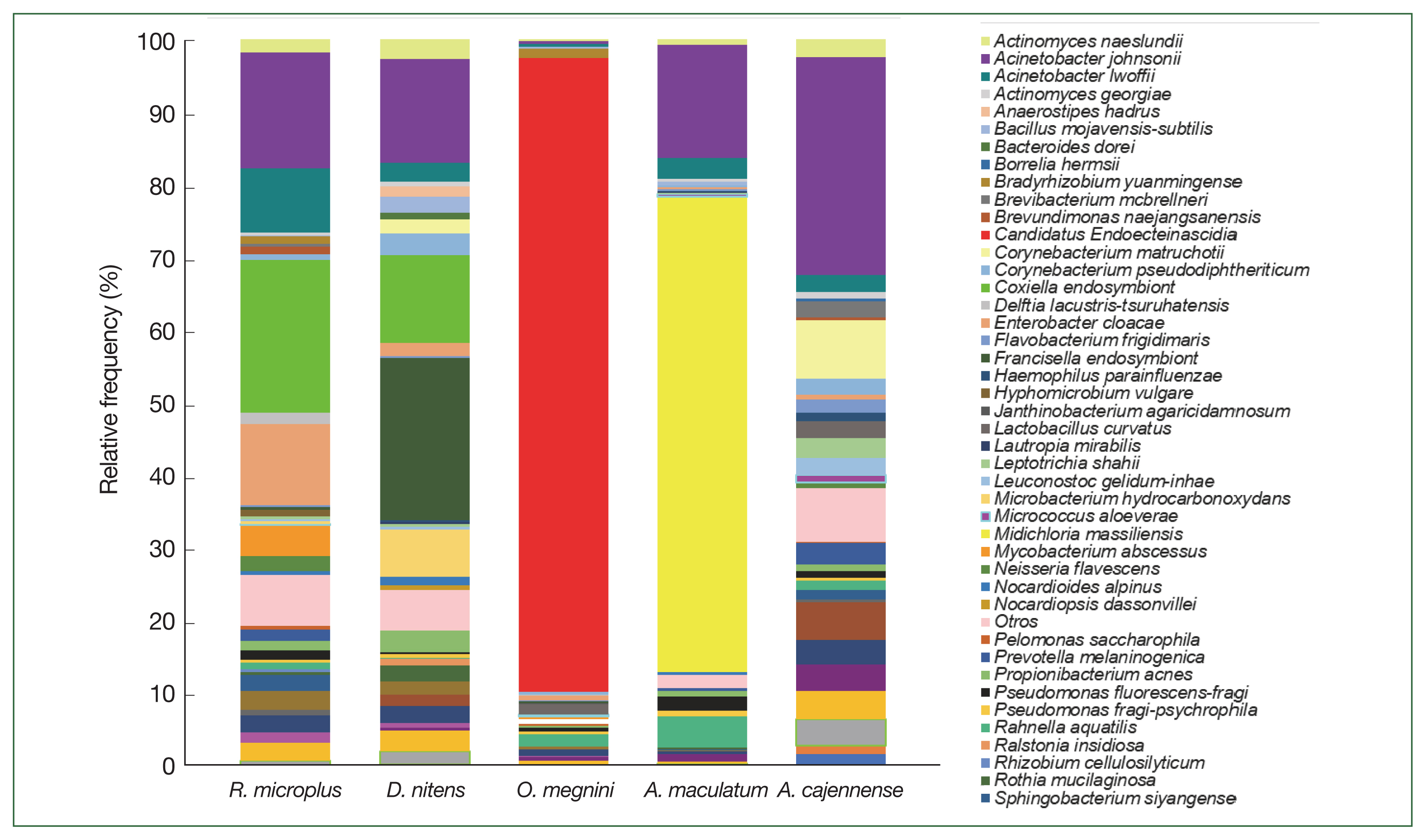
Fig. 6Plot of beta diversities of the microbiomes of Rm (Coah and Chih), Dn (Chih and NL), and Am (Coah); clustered separately from Rm NL F, Rm Tam F, and Ac NL M. Principal coordinates (PCo1 and PCo2) depicting differences in the taxonomic composition of the bacterial communities based on Bray-Curtis indices. The samples inside the ellipses are clustered separately from the remaining groups(95% confidence intervals).
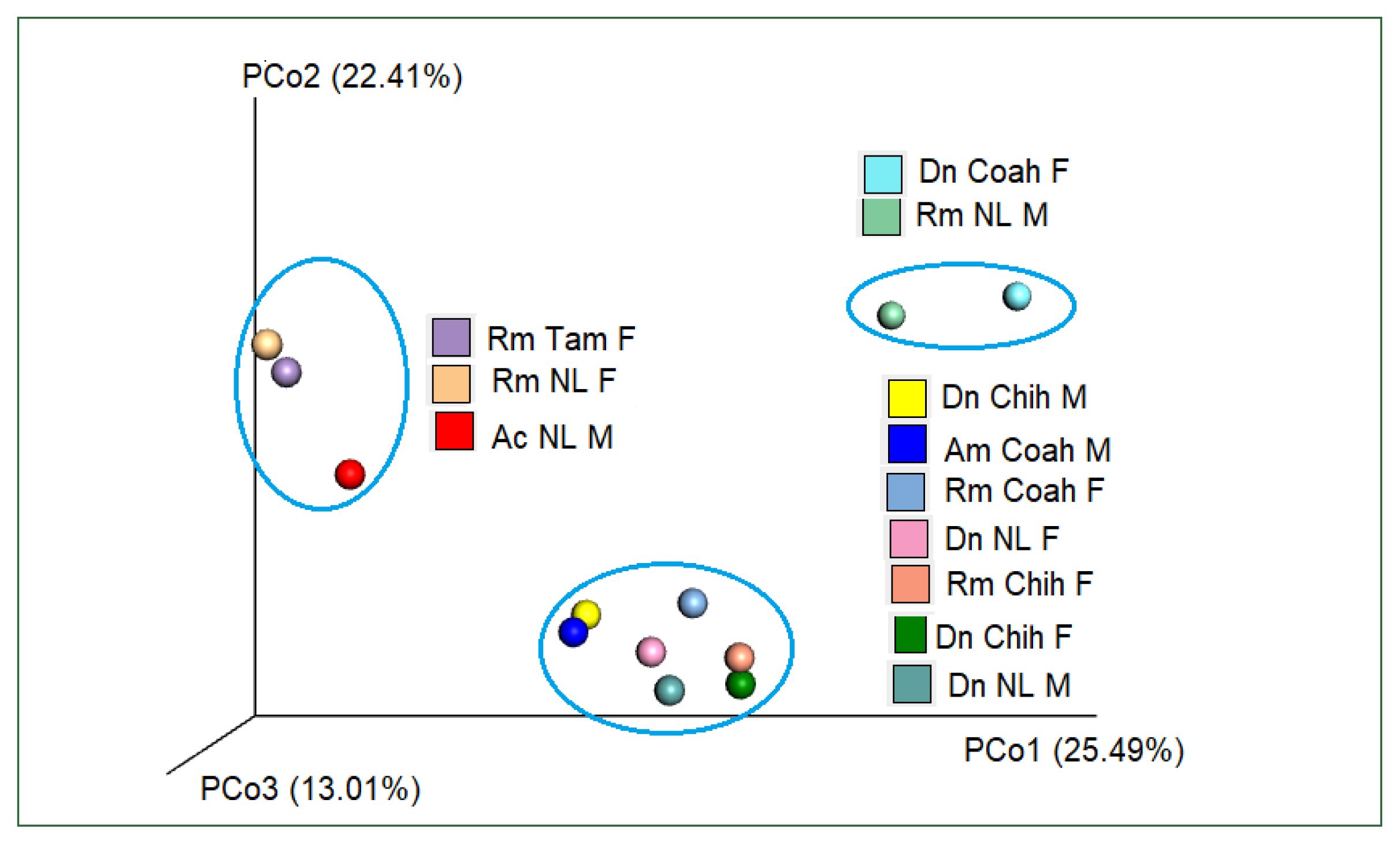
Table 1Ticks collected from white-tailed deer (Odocoileus virginianus) in 5 states located in northern Mexico
Table 1
|
Locality (Geographic coordinates) |
R. microplus
|
D. nitens
|
A. cajennense
|
A. maculatum
|
O. megnini
|
Infested O. virginianus
|
|
|
|
|
|
|
F |
M |
F |
M |
F |
M |
F |
M |
Nymph |
|
Salinas Victoria, Nuevo Leon (26°7′59.88″ N, 100°19′58.8″ W) |
87 |
14 |
30 |
39 |
0 |
6 |
0 |
0 |
0 |
32 |
|
|
Nuevo Laredo, Tamaulipas (27°33′0″ N, 99°47′59.9″ W) |
17 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
6 |
|
|
Muzquiz, Coahuila (28°7′0.12″ N, 101°46′ 58.8″ W) |
10 |
0 |
27 |
5 |
0 |
0 |
0 |
2 |
0 |
8 |
|
|
Casas Grandes, Chihuahua (30°23′44.99″ N, 108°12′ 30″ W) |
12 |
0 |
17 |
5 |
0 |
0 |
0 |
0 |
0 |
5 |
|
|
La Colorada, Sonora (28°43′41″ N, 110°26′ 5″ W) |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
0 |
19 |
6 |
|
|
Total |
126 |
14 |
74 |
49 |
0 |
6 |
0 |
2 |
19 |
57 |
Table 2Raw sequences and number of OTUs obtained from tick species collected from legal white-tailed deer game farms in 5 states of northern Mexico
Table 2
|
Tick species |
Locality/sex |
Raw sequences (R1+R2) |
Chimeric sequences |
No. of OTUs (identified species) |
SRA Accession no. |
|
R. microplus
|
Coah F |
106,160 |
6,295 |
36 |
SRX18276467 |
|
Tam F |
89,234 |
313 |
86 |
SRX18276466 |
|
NL F |
123,552 |
66 |
42 |
SRX17608462 |
|
NL M |
45,656 |
0 |
11 |
SRX17608463 |
|
Chih F |
69,886 |
1,828 |
30 |
SRX17608463 |
|
|
D. nitens
|
Chih F |
47,584 |
232 |
18 |
SRX18276468 |
|
Chih M |
37,508 |
122 |
57 |
SRX18276469 |
|
NL F |
91,710 |
1,204 |
31 |
SRX17608464 |
|
NL M |
91,912 |
25 |
32 |
SRX17608465 |
|
Coah F |
160,916 |
9,021 |
18 |
SRX17608466 |
|
Coah M |
2,172 |
2 |
19 |
SRX17608467 |
|
|
A. maculatum
|
Coah M |
42,020 |
551 |
58 |
SRX17608468 |
|
|
A. cajennense
|
NL M |
32,838 |
1,016 |
69 |
SRX17608469 |
|
|
O. megnini
|
Son N |
94,564 |
16 |
30 |
SRX18275422 |
|
|
Total |
1,035,712 |
20,691 |
537 |
|
Table 3SIMPER analysis between tick species. The 5 microbial families with the highest percentage of contribution are shown
Table 3
|
R. microplus
|
A. maculatum
|
A. cajennense
|
O. megnini
|
|
|
|
|
|
Taxon (Family) |
Contribution (%) |
Taxon (Family) |
Contribution (%) |
Taxon (Family) |
Contribution (%) |
Taxon (Family) |
Contribution (%) |
|
D. nitens
|
average dissimilarity: 49.8 |
average dissimilarity: 72.9 |
average dissimilarity: 56.6 |
average dissimilarity: 91.8 |
|
Francisellaceae |
23.5 |
Midichloriaceae |
46.5 |
Francisellaceae |
21.0 |
Thiotrichales |
49.0 |
|
Enterobacteriaceae |
10.7 |
Francisellaceae |
15.8 |
Moraxellaceae |
14.7 |
Francisellaceae |
12.3 |
|
Coxiellaceae |
9.61 |
Coxiellaceae |
8.5 |
Coxiellaceae |
11.3 |
Moraxellaceae |
8.9 |
|
Moraxellaceae |
8.54 |
Microbacteriaceae |
4.5 |
Microbacteriaceae |
6 |
Coxiellaceae |
6.7 |
|
Microbacteriaceae |
6.4 |
Corynebacteriaceae |
3.4 |
Corynebacteriaceae |
4.9 |
Microbacteriaceae |
3.5 |
|
|
R. microplus
|
|
|
average dissimilarity: 67.6 |
average dissimilarity: 52.4 |
average dissimilarity: 89.4 |
|
|
Midichloriaceae |
50.5 |
Coxiellaceae |
21.5 |
Thiotrichales |
50.6 |
|
|
Coxiellaceae |
16.2 |
Enterobacteriaceae |
10.3 |
Moraxellaceae |
13.9 |
|
|
Enterobacteriaceae |
5.7 |
Corynebacteriaceae |
9.6 |
Coxiellaceae |
12.1 |
|
|
Moraxellaceae |
4.8 |
Moraxellaceae |
7.7 |
Enterobacteriaceae |
5.7 |
|
|
Mycobacteriaceae |
3.3 |
Staphylococcaceae |
6.3 |
Mycobacteriaceae |
2.2 |
|
|
A. maculatum
|
|
|
|
|
average dissimilarity: 70.9 |
average dissimilarity: 92.8 |
|
|
|
|
Midichloriaceae |
48.3 |
Thiotrichales |
47.5 |
|
|
|
|
Moraxellaceae |
10.2 |
Midichloriaceae |
35.6 |
|
|
|
|
Corynebacteriaceae |
7.3 |
Moraxellaceae |
9.6 |
|
|
|
|
Staphylococcaceae |
6.05 |
Enterobacteriaceae |
1.2 |
|
|
|
|
Streptococcaceae |
4.5 |
Pseudomonadaceae |
0.9 |
|
|
A. cajennense
|
|
|
|
|
|
|
average dissimilarity: 90.3 |
|
|
|
|
|
|
Thiotrichales |
50.2 |
|
|
|
|
|
|
Moraxellaceae |
18.1 |
|
|
|
|
|
|
Corynebacteriaceae |
5.6 |
|
|
|
|
|
|
Staphylococcaceae |
4.4 |
|
|
|
|
|
|
Streptococcaceae |
3.6 |
References
- 1. Bonnet SI, Binetruy F, Hernández-Jarguín AM, Duron O. The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front Cell Infect Microbiol 2017;7:1-14. https://doi.org/10.3389/fcimb.2017.00236
- 2. Kaufman EL, Stone NE, Scoles GA, Hepp CM, Busch JD, et al. Range-wide genetic analysis of Dermacentor variabilis and its Francisella-like endosymbionts demonstrates phylogeographic concordance between both taxa. Parasit Vectors 2018;11(1):306. https://doi.org/10.1186/s13071-018-2886-5
- 3. Landesman WJ, Mulder K, Fredericks LP, Allan BF. Cross-kingdom analysis of nymphal-stage Ixodes scapularis microbial communities in relation to Borrelia burgdorferi infection and load. FEMS Microbiol Ecol; 2019. 95(12):fiz167 https://doi.org/10.1093/femsec/fiz167
- 4. Greay TL, Gofton AW, Paparini A, Ryan UM, Oskam CL, et al. Recent insights into the tick microbiome gained through next-generation sequencing. Parasit Vectors 2018;11:1-14. https://doi.org/10.1186/s13071-017-2550-5
- 5. Paddock CD, Yabsley MJ. Ecological havoc, the rise of white-tailed deer, and the emergence of Amblyomma americanum-associated zoonoses in the united states. In Childs JE, Mackenzie JS, Richt JA eds, Wildlife and Emerging Zoonotic Diseases: the Biology, Circumstances and Consequences of Cross-Species Transmission. Springer; Heidelberg, Germany. 2007, 315:pp 289-324 https://doi.org/10.1007/978-3-540-70962-6_12
- 6. Medrano C, Boadella M, Barrios H, Cantú A, García Z. Zoonotic pathogens among white-tailed deer, northern Mexico, 2004–2009. Emerg Infect Dis 2012;18(8):1372-1374. https://doi.org/10.3201/eid1808.111902
- 7. Cantú-Martinez M, Salinas-Meléndez JA, Zarate-Ramos J, Ávalos-Ramírez R, Martínez-Muñoz , et al. Prevalence of antibodies against Babesia bigemina and B. bovis in white-tailed deer (Odocoileus virginianus texanus) in farms of northeastern Mexico. J Anim Vet Adv 2008;7(2):121-123.
- 8. Martinez A, Salinas A, Martinez F, Cantu A, Miller DK. Serosurvey for selected disease agents in white-tailed deer from Mexico. J Wildl Dis 1999;35(4):799-803. https://doi.org/10.7589/0090-3558-35.4.799
- 9. Instituto Nacional de Estadística y Geografia. 2024. Espacio y datos de México; https://www.inegi.org.mx/app/mapa/espacioydatos/
- 10. Hoskins JD. Ixodid and argasid ticks. Keys to their identification. Vet Clin North Am Small Anim Pract 1991;21:185-197. http://doi.org/10.1016/s0195-5616(91)50018-8
- 11. Lucero-Velasco EA, Molina-Garza ZJ, Galaviz-Silva L. First survey of cultivable bacteria from Rhipicephalus sanguineus sensu lato and assessment of the antagonism against five microorganisms of clinical importance assessment of the antagonism against five microorganisms of clinical importance. Int J Acarol 2018;44(4–5):204-209. https://doi.org/10.1080/01647954.2018.1495262
- 12. Molina-Garza ZJ, Galaviz-Silva L. Cultivable bacteria isolated from cattle ticks of nuevo León and Zacatecas, Mexico, and an assessment of their antagonism against bacteria of clinical concern. Int J Acarol 2019;46(1):1-7. https://doi.org/10.1080/01647954.2019.1696405
- 13. Li LH, Zhang Y, Zhu D. Effects of antibiotic treatment on the fecundity of Rhipicephalus haemaphysaloides ticks. Parasit Vectors 2018;11(1):1-7. https://doi.org/10.1186/s13071-018-2807-7
- 14. Callahan BJ, Mcmurdie PJ, Rosen MJ, Han A, Johnson JA, et al. DADA2: high resolution sample inference from Illumina amplicon data. Nat Methods 2016;13(7):581-583. https://doi.org/10.1038/nmeth.3869.DADA2
- 15. Caporaso GJ, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7(5):335-336. https://doi.org/10.1038/nmeth.f.303
- 16. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12(6):R60. https://doi:10.1186/gb-2011-12-6-r60
- 17. Sayler KA, Loftis AD, Beatty SK, Boyce CL, Garrison E, et al. Prevalence of tick-borne pathogens in host-seeking Amblyomma Americanum (Acari: Ixodidae) and Odocoileus virginianus (Artiodactyla: Cervidae) in Florida. J Med Entomol 2016;53(4):949-956. https://doi.org/10.1093/jme/tjw054
- 18. Vor T, Kiffner C, Hagedorn P, Niedrig M, Rühe F. Tick burden on European roe deer (Capreolus capreolus). Exp Appl Acarol 2010;51(4):405-417. https://doi.org/10.1007/s10493-010-9337-0
- 19. Diyes GCP, Rajakaruna RS. Seasonal dynamics of spinose ear tick Otobius megnini associated with horse otoacariasis in sri lanka. Acta Trop 2016;159:170-175. https://doi.org/10.1016/j.actatropica.2016.03.025
- 20. Kellogg FE, Kistner TP, Strickland RK, Gerrish RR. Arthropod parasites collected from white-tailed deer. J Med Entomol 1971;8(5):495-498.
- 21. Segura JA, Isaza JP, Botero LE, Alzate JF, Gutiérrez LA. Assessment of bacterial diversity of Rhipicephalus microplus ticks from two livestock agroecosystems in antioquia, colombia. PLoS One 2020;15(7):e0234005. https://doi.org/10.1371/journal.pone.0234005
- 22. Ojeda-Chi MM, Rodriguez-Vivas RI, Esteve-Gasent MD, de León AP, Modarelli JJ, et al. Molecular detection of rickettsial tick-borne agents in white-tailed deer (Odocoileus virginianus yucatanensis), mazama deer (Mazama temama), and the ticks they host in Yucatan, Mexico. Ticks Tick Borne Dis 2019;10(2):365-370. https://doi.org/10.1016/j.ttbdis.2018.11.018
- 23. Hernández-Jarguín A, Díaz-Sánchez S, Villar M, de la Fuente J. Integrated metatranscriptomics and metaproteomics for the characterization of bacterial microbiota in unfed Ixodes ricinus. Ticks Tick Borne Dis 2018;9(5):1241-1251. https://doi.org/10.1016/j.ttbdis.2018.04.020
- 24. Clayton KA, Gall CA, Mason KL, Scoles GA, Brayton KA. The characterization and manipulation of the bacterial microbiome of the Rocky Mountain wood tick, Dermacentor andersoni. Parasit Vectors 2015;8:1-5. https://doi.org/10.1186/s13071-015-1245-z
- 25. Narasimhan S, Fikrig E. Tick microbiome: the force within. Trends Parasitol 2015;31(7):315-323. https://doi.org/10.1016/j.pt.2015.03.010
- 26. Scoles GA. Phylogenetic analysis of the Francisella-like endosymbiont of Dermacentor ticks. J Med Entomol 2004;41(3):277-286. https://doi.org/10.1603/0022-2585-41.3.277
- 27. Guizzo MG, Tirloni L, Gonzalez SA, Farber MD, Braz G, et al. Coxiella Endosymbiont of Rhipicephalus microplus modulates tick physiology with a major impact in blood feeding capacity. Front Microbiol 2022;13:868575. https://doi:10.3389/fmicb.2022.868575
- 28. Moutailler S, Valiente Moro C, Vaumourin E, Michelet L, Tran FH, et al. Co-infection of ticks: the rule rather than the exception. PLoS Negl Trop Dis 2016;10(3):1-17. https://doi.org/10.1371/journal.pntd.0004539
- 29. Vayssier-Taussat M, Kazimirova M, Hubalek Z, Hornok S, Farkas R, et al. Emerging horizons for tick-borne pathogens: from the “one pathogen-one disease” vision to the pathobiome paradigm. Future Microbiol 2015;10(12):2033-2043. https://doi.org/10.2217/fmb.15.114
- 30. Venzal JM. Epidemiology of rickettsioses by Rickettsia parkeri andother emerging and reemerging species associated with anthropization in Latin America. Acta Méd Costarric 2013;55(suppl 1):45-47. (in Spanish).
- 31. Gofton AW, Oskam CL, Lo N, Beninati T, Wei H, et al. Inhibition of the endosymbiont “Candidatus Midichloria mitochondrii” during 16S rRNA gene profiling reveals potential pathogens in Ixodes ticks from Australia. Parasit Vectors 2015;8:1-11. https://doi.org/10.1186/s13071-015-0958-3
- 32. Sakamoto JM, Silva-Diaz GE, Wagner EA. Microbial ecology of Ixodes scapularis from Central Pennsylvania, USA. bioRxiv 2020;197335. https://doi.org/10.1101/2020.07.11.197335
- 33. Rojas-Jaimes J, Lindo-Seminario D, Correa-Núñez G, Diringer B. Characterization of the bacterial microbiome of Rhipicephalus (Boophilus) microplus collected from Pecari tajacu “Sajino” Madre de Dios, Peru. Sci Rep 2021;11(1):6661. https://doi.org/10.1038/s41598-021-86177-3
- 34. Ponnusamy L, Gonzalez A, Van Treuren W, Weiss S, Parobek CM, et al. Diversity of rickettsiales in the microbiome of the lone star tick, Amblyomma americanum. Appl Environ Microbiol 2014;80(1):354-359. https://doi.org/10.1128/AEM.02987-13
- 35. Travanty N, Ponnusamy L, Kakumanu ML, Nicholson WL, Apperson CS. Diversity and structure of the bacterial microbiome of the American dog tick, Dermacentor variabilis, is dominated by the endosymbiont Francisella.. Symbiosis 2019;79:239-250. https://doi.org/10.1007/s13199-019-00642-2
- 36. Telford III SR. Status of the “East side hypothesis” (transovarial interference) twenty five years later. Ann NY Acad Sci 2009;1166:144-150. https://doi.org/10.1111/j.1749-6632.2009.04522.x

 , Mariana Cuesy-León1
, Mariana Cuesy-León1