Abstract
Acanthamoeba is an opportunistic pathogen that causes Acanthamoeba keratitis, granulomatous amoebic encephalitis, and other cutaneous diseases. The life cycle of Acanthamoeba consists of 2 stages of trophozoites and cysts. Under adverse environmental conditions, Acanthamoeba encysts, while the conditions become favorable for growth, it reverts to the trophozoite form. Acanthamoeba excystation is crucial for its proliferation and can lead to recurrent infections after incomplete treatment. To identify the factors involved in excystation, A. castellanii was subjected to either encystation- or excystation-inducing conditions, and gene expression profiles were compared using mRNA sequencing. A. castellanii samples were collected at 8 h intervals for analysis under both conditions. Differentially expressed gene analysis revealed that 1,214 and 1,163 genes were upregulated and downregulated, respectively, by more than 2-fold during early excystation. Five genes markedly upregulated in early excystation (ACA1_031140, ACA1_032330, ACA1_374400, ACA1_275740, and ACA1_112650) were selected, and their expression levels were confirmed via real-time PCR. Small interfering RNA (siRNA) targeting these 5 genes was transfected into Acanthamoeba and gene knockdown was validated through real-time PCR. The silencing of ACA1_031140, ACA1_032330, ACA1_374400, and ACA1_112650 inhibited excystation and suggested that these genes might be essential for excystation. Our findings provide valuable insights for suppressing Acanthamoeba proliferation and recurrence.
-
Key words: Acanthamoeba, encystation, excystation, gene knockdown
Introduction
Acanthamoeba spp. are opportunistic pathogens and causative agents of severe infections, including
Acanthamoeba keratitis and granulomatous amoebic encephalitis [
1]. The life cycle of
Acanthamoeba spp. included such 2 stages as the vegetative trophozoite and resilient cyst form [
2]. Under favorable growth conditions, the trophozoite stage predominates, but when faced with adverse conditions, such as nutrient deprivation, pH fluctuations, or drug exposure, the trophozoite transitions into the cyst form [
3].
Acanthamoeba cyst form has garnered attention owe to its resistance to various biocides and disinfectants [
4]. Additionally, cysts can remain viable and pathogenic for several years, play a key role in transmitting
Acanthamoeba infections [
5]. Consequently, advancements in antimicrobial agents have targeted the cyst stage or aimed to prevent encystation. Despite these advances, recurrent
Acanthamoeba infections caused by the excystation of drug-resistant cysts remain a persistent challenge [
6].
Excystation, where cysts revert to the metabolically active trophozoite form, is critical to both the onset and recurrence of
Acanthamoeba infections [
7]. Although encystation and amebic drug resistance have been extensively researched, there remains a lack of studies on amebic excystation.
Acanthamoeba cysts possess pores known as ostioles, through which they monitor environmental changes. Under favorable conditions, trophozoites emerge from the cysts through these ostioles, initiating excystation. Previous studies have elucidated the morphological changes that occur during excystation as well as the effects of various environmental and physiological conditions on
A. castellanii excystation [
7,
8]. Nonetheless, genetic studies on excystation are scarce. Specifically, no comprehensive analysis of gene expression changes during excystation has been reported. Given that the transitions between trophozoites and cysts involve different gene expression patterns, understanding how these mechanisms are genetically regulated could improve
Acanthamoeba keratitis treatment.
To address this knowledge gap, we investigated the total transcriptional changes of A. castellanii during excystation (8, 16, 24, and 48 h) using RNA sequencing, comparing them to the encystation process (8, 16, 24, and 48 h). Furthermore, several highly expressed genes during early excystation were identified, and their potential roles in this process were explored. Our findings may provide a foundation for developing more effective treatments for Acanthamoeba infections.
Materials and Methods
Ethics statement
Not applicable.
Cultivation, encystation, and excystation of Acanthamoeba
Acanthamoeba castellanii (ATCC 30868) was obtained from the American Type Culture Collection and cultured axenically in PYG medium (20 g/L proteose peptone, 1 g/L yeast extract, 0.1 M glucose, 4 mM MgSO4, 0.4 mM CaCl2, 3.4 mM sodium citrate, 0.05 mM Fe(NH4)2(SO4)2, 2.5 mM Na2HPO4, and 2.5 mM KH2PO4 (pH 6.5)) at 25°C. Encystation was induced using encystment medium (95 mM NaCl, 5 mM KCl, 8 mM MgSO4, 0.4 mM CaCl2, 1 mM NaHCO3, and 20 mM Tris-HCl (pH 9.0)) at 25°C. Excystation was induced on a 1.5% non-nutrient agar (Thermo Fisher, Waltham, MA, USA) plate coated with heat-treated Escherichia coli. Morphological changes during encystation and excystation were observed using a light microscope.
RNA isolation, library preparation, and sequencing data analysis
Total RNA was isolated from
Acanthamoeba trophozoites, encysting
Acanthamoeba (8, 16, 24, and 48 h), and excysting
Acanthamoeba (8, 16, 24, and 48 h) using the RNeasy Mini Kit (Qiagen, Hilden, Germany). All subsequent RNA sequencing steps were performed by eBiogen (Seoul, Korea). Libraries were prepared from 2 μg of total RNA using the SMARTer Stranded RNA-Seq Kit (Clontech Laboratories, Mountain View, CA, USA). mRNA was isolated using the Poly(A) RNA Selection Kit (Lexogen, Vienna, Austria). Isolated mRNA was used for cDNA synthesis and shearing, following the manufacturer’s instructions. Indexing was performed using Illumina indexes 1–12, and PCR was used for enrichment. Library quality was assessed using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) to evaluate mean fragment size, and quantification was performed using a library quantification kit on a StepOne Real-Time PCR System (Thermo Fisher Scientific, Branford, CT, USA). High-throughput sequencing was performed as paired-end 100 sequencing using the HiSeq 2500 system (Illumina, San Diego, CA, USA). For sequencing analysis, mRNA sequencing reads were mapped using TopHat to obtain alignment files. Differentially expressed genes (DEGs) were identified based on counts from unique and multiple alignments using coverage in Bedtools. Read count data were normalized using the quantile normalization method in EdgeR within R using Bioconductor. The alignment files were also employed to assemble transcripts, estimate abundances, and detect differential gene or isoform expression using Cufflinks. Gene expression levels were calculated using the fragments per kilobase of exon per million fragments method. Genes were classified based on DAVID searches (
http://david.abcc.ncifcrf.gov/).
Total RNA was purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized using the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Hanover, IN, USA). Real-time PCR was performed on a Magnetic Induction Cycler PCR system (PhileKorea, Seoul, Korea) using the following default thermocycler program for all genes: preincubation at 95°C for 1 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec. Reaction mixtures were prepared using Luna Universal qPCR Master Mix (New England Biolabs, Ipswich, MA, USA) with different sense and antisense primers (
Supplmentary Table S1). The reference gene was 18S ribosomal DNA.
Small interfering RNAs (siRNAs) targeting 5
Acanthamoeba genes (
ACA1_031140,
ACA1_ 032330,
ACA1_374400,
ACA1_275740, and
ACA1_112650) were synthesized by Bioneer Inc. (Bioneer, Daejeon, Korea) based on their cDNA sequences (
Supplmentary Table S2). The siRNA (final concentration: 100 nM) was transfected into live
Acanthamoeba trophozoites at a cell density of 4×10
5 cells using Effectene transfection reagent (Qiagen, Hilden, Germany) following the manufacturer’s protocol. Transfection efficiency was determined by observing fluorescing cells under a fluorescent microscope (Leica, Wetzlar, Germany). siRNA-transfected
Acanthamoeba underwent encystation for 48 h, followed immediately by excystation for 8 h. Real-time PCR was then performed as described above. The Effectene transfection reagent was used as a negative control.
Data are presented as the means±standard deviations (SD) from 3 independent experiments. Statistical analyses were performed using Student’s t-test. P-values >0.05 were considered statistically significant.
Results
DEGs of Acanthamoeba during encystation and excystation
To identify genes involved in
Acanthamoeba excystation, DEGs during encystation (8, 16, 24, and 48 h) and excystation (8, 16, 24, and 48 h) were compared using mRNA sequencing. A total of 15,655 genes in
Acanthamoeba exhibited more than a 2-fold change in expression during these processes (
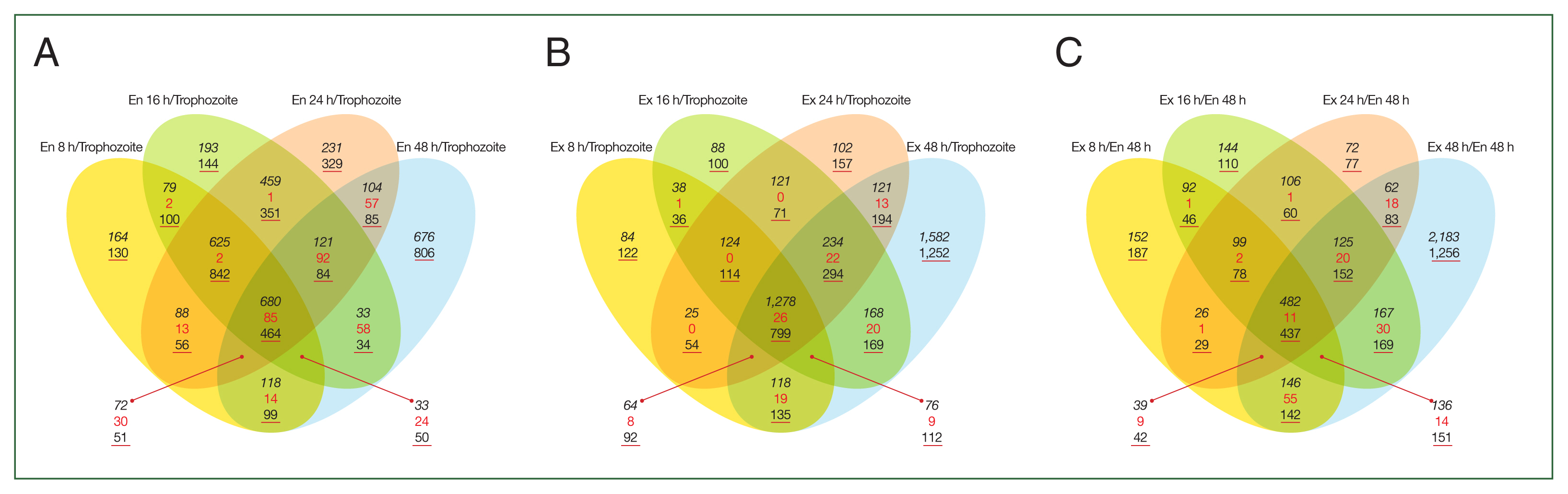
Table 1). A Venn diagram revealed that some DEGs overlapped between the encystation or excystation time-course (8, 16, 24, and 48 h) groups (
Fig. 1). The expression of numerous genes was confirmed to either increase or decrease during both stages. We focused on genes highly expressed during the early stages of excystation (after 8 h of excystation) in mature
Acanthamoeba cysts (after 48 h of encystation).
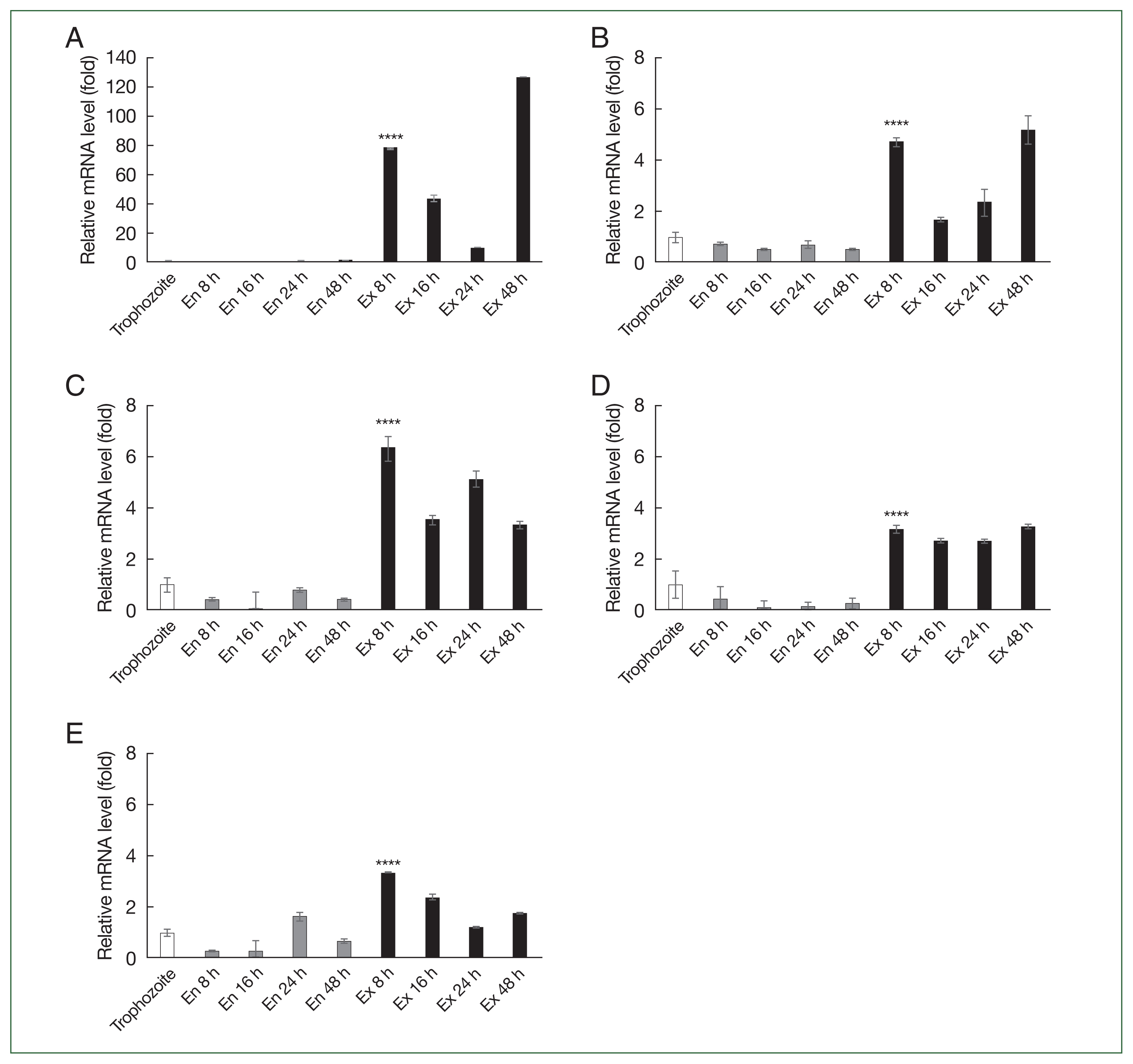
Based on mRNA sequencing analysis, 5 DEGs (
ACA1_031140,
ACA1_032330,
ACA1_374400,
ACA1_275740, and
ACA1_112650) highly expressed during the early stages of excystation compared to encystation (Ex 8/En 48) were selected for validation via real-time quantitative PCR. The genes and their primers are listed in
Supplementary Table S1. As shown in
Fig. 2, none of the 5 genes showed increased expression during encystation, but their expression increased during excystation. Specifically,
ACA1_031140 showed an approximately 80-fold increase in expression 8 h into excystation (
Fig. 2A). At 48 h after excystation, many trophozoites (the active feeding stage of amoebae) were already present, indicating that excystation was likely complete; therefore, this stage may not form part of the ongoing excystation process. Thus, the significant increase in gene expression at 48 h after excystation was not considered meaningful (
Fig. 2A).
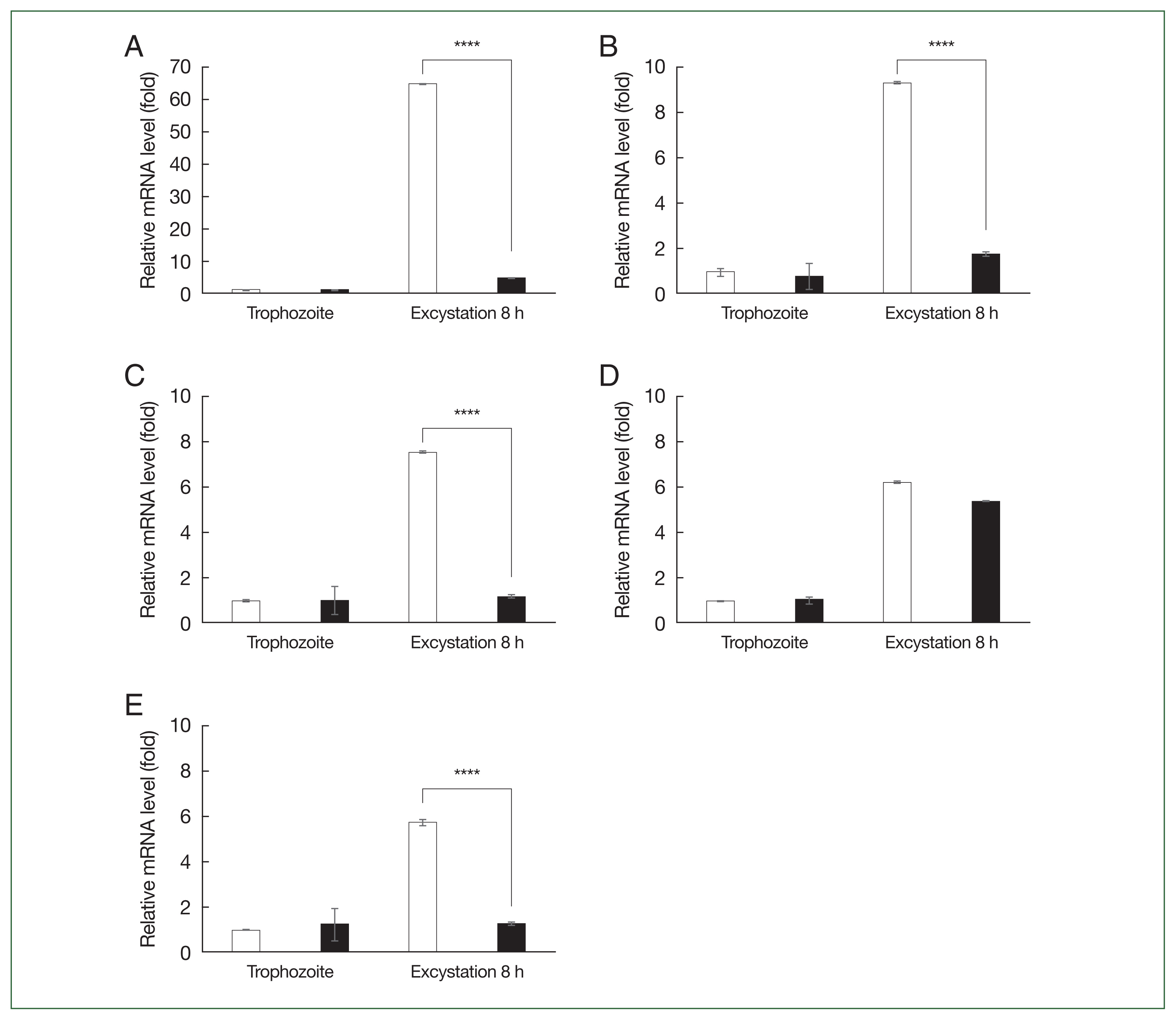
To observe the effects of gene knockdown in vitro, 5 siRNAs specific to
ACA1_031140,
ACA1_ 032330,
ACA1_374400,
ACA1_275740, and
ACA1_112650 were prepared (
Supplementary Table S2). After transfecting
Acanthamoeba with these siRNAs, real-time PCR was performed to evaluate gene silencing efficiency (
Fig. 3). In the trophozoite control groups, siRNA transfection did not affect mRNA levels (
Fig. 2A–
E). However, in the 8 h excystation group, siRNA transfection significantly reduced mRNA expression of
ACA1_031140,
ACA1_ 032330,
ACA1_374400, and
ACA1_112650 to basal levels (
Fig. 2A–
C and
E). Transfecting siRNA specific to
ACA1_275740 had a negligible impact on the 8 h excystation group (
Fig. 2D).
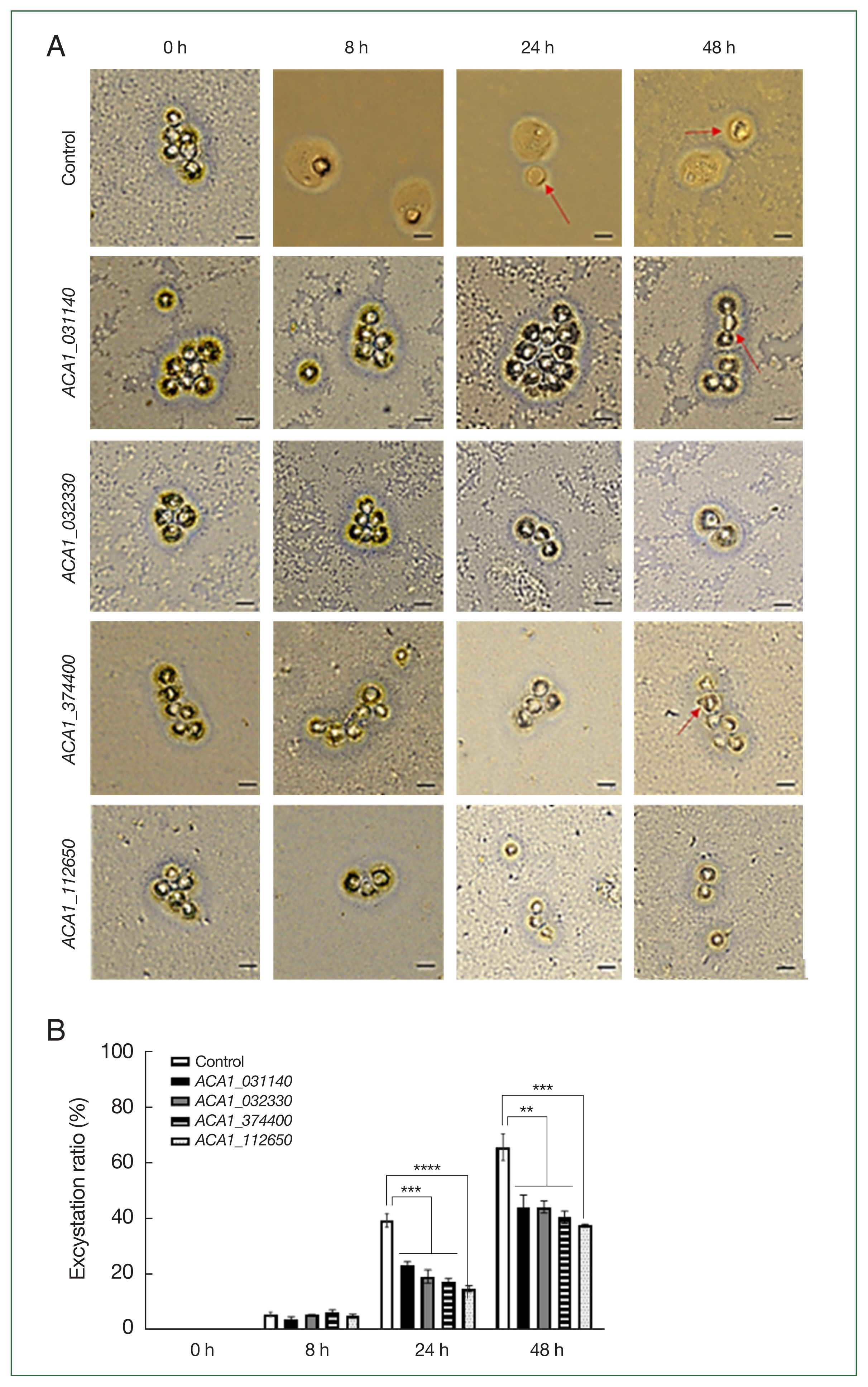
To investigate the effects of
ACA1_031140,
ACA1_032330,
ACA1_374400, and
ACA1_ 112650 gene knockdown in
Acanthamoeba, the excystation ratio was examined (
Fig. 4). The excystation rate was calculated by counting trophozoites and cyst shells that emerged after inducing encystation of siRNA-transfected amoebae and culturing them on an agar plate coated with heat-treated
E. coli (
Fig. 4A, red arrows indicate empty cysts). The encystation rate shown in
Fig. 4A was quantified and presented in
Fig. 4B. After inducing excystation in
Acanthamoeba mature cysts, excystation rates were 6.3±0.9%, 40.0±2.7%, and 65.9±5.2% at 8, 24, and 48 h, respectively. In siRNA-treated
Acanthamoeba, no significant differences in excystation rates were observed at 8 h compared to the control group. However, as time progressed, significant differences appeared, with excystation rates reaching 24.0±0.7%, 17.6±1.3%, 18.5±1.2%, and 14.9±1.1% at 24 h for
ACA1_031140,
ACA1_ 032330,
ACA1_374400, and
ACA1_112650, respectively (
Fig. 4B). By 48 h, excystation rates were 43.6±4.9%, 43.6±1.5%, 41.0±2.3%, and 37.4±0.5% for the 4 aforementioned genes, respectively, indicating a 20% reduction in excystation rates. Although not all time points showed significant differences in excystation rates for siRNA-treated groups, the lowest excystation rate was observed with
ACA1_112650 inhibition.
Discussion
Encystation and excystation are critical processes in the life cycle of
Acanthamoeba spp., playing key roles in survival, drug resistance, and infection. Understanding the molecular mechanisms underlying these processes could reveal potential targets for disrupting
Acanthamoeba’s life cycle. In this study, we identified 15,655 DEGs involved in encystation and excystation in
A. castellanii, finding 4 genes that suppressed excystation by 20% during the process’ early stages. BLAST analysis revealed that DEGs
ACA1_031140,
ACA1_032330, and
ACA1_374400 encode hypothetical proteins with unknown functions, whereas
ACA1_ 362260 encodes nicotinamide N-methyltransferase (NNMT;
Supplementary Table S1), a metabolic enzyme involved in regulating methylation potential, which in turn affects DNA and histone epigenetic modifications [
9]. NNMT plays a major role in the development of various cancers and shows promise as a biomarker for tumor diagnosis and treatment [
10]. Although NNMT’s role in
Acanthamoeba spp. remains unreported, its increased expression during early excystation suggests that it may regulate the expression of genes critical for excystation.
During encystation in
Acanthamoeba spp., rounded trophozoites appear approximately 12 h after induction of encystation [
11]. Immature precysts with a continuous cell wall form around 24 h later, with the number of precysts increasing such that by 72 h, 70% of the trophozoites develop into mature cysts with a characteristic double-layered cyst wall. Time-lapse, phase-contrast studies on excystation have shown that the amoeba detaches from the inner cyst wall within 4–8 h after excystation induction [
12]. Between 8 and 24 h, the detachment becomes more pronounced, and cytoplasmic buds penetrate the cyst wall, eventually leading to the emergence of a trophozoite, leaving behind an empty cyst wall. Based on these observations, gene expression changes were monitored at 8, 16, 24, and 48 h during encystation and excystation (
Table 1;
Fig. 1). We posited that genes showing increased expression during early excystation in
Acanthamoeba play pivotal roles in this process. Therefore, we focused on 5 genes for which expression increased 8 h after excystation began:
ACA1_031140,
ACA1_032330,
ACA1_374400,
ACA1_275740, and
ACA1_112650.
In conclusion, this study identified numerous genes, that exhibit expression changes during encystation and excystation (
Table 1). However, owing to the limited genomic information available for
Acanthamoeba spp., many DEGs were found to encode hypothetical proteins, complicating efforts to characterize their functions. Further research is required to explore and characterize these DEGs across different stages of
Acanthamoeba’s life cycle. In particular, focusing on the 4 excystation-related genes identified in this study could provide insights into their function and shared mechanisms among pathogenic amoebae. Such findings could contribute to developing improved treatment and prevention strategies for
Acanthamoeba infections and combating other cyst-forming protozoa.
Notes
-
Author contributions
Conceptualization: Kong HH, Moon EK
Data curation: Kim MJ, Jo HJ, Quan FS, Chu KB, Moon EK
Funding acquisition: Moon EK
Investigation: Kim MJ, Jo HJ, Moon EK
Methodology: Kim MJ, Jo HJ, Quan FS, Chu KB, Kong HH, Moon EK
Writing – original draft: Moon EK
Writing – review & editing: Kim MJ, Jo HJ, Quan FS, Chu KB, Kong HH
-
The authors declare no conflict of interest related to this study.
Supplementary Information
Acknowledgement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (RS-2024-00346635).
Fig. 1Venn diagram depicting the analysis of differentially expressed genes over time during the encystation and excystation of A. castellanii. (A) Encystation 8, 16, 24, and 48 h compared to trophozoites. (B) Excystation 8, 16, 24, and 48 h compared to trophozoites. (C) Excystation 8, 16, 24, and 48 h compared to encystation at 48 h.

Fig. 2Real-time PCR analysis of differentially expressed genes (DEGs) in A. castellanii. mRNA expression levels of 5 DEGs in A. castellanii during encystation and excystation were assessed using real-time PCR. (A) ACA1_031140. (B) ACA1_032330. (C) ACA1_374400. (D) ACA1_275740. (E) ACA1_112650. The transcription levels of these 5 genes were compared among Acanthamoeba trophozoites and encysting and excysting forms. Data are shown as the mean±SD, representing 3 independent experiments. Asterisks indicate statistical significance: ****P<0.0001 compared to Acanthamoeba trophozoites.

Fig. 3Real-time PCR analysis following siRNA transfection. Acanthamoeba castellanii were transfected with siRNAs targeting the 5 DEGs. After 48 h of encystation, excystation was immediately induced for 8 h, and gene knockdown was examined. (A) ACA1_031140. (B) ACA1_032330. (C) ACA1_374400. (D) ACA1_275740. (E) ACA1_112650. Data are presented as the mean±SD, representative of 3 independent experiments. ****P<0.0001 between the control group (□) and the siRNA-transfected group (■).

Fig. 4Evaluation of excystation in siRNA-transfected Acanthamoeba. After siRNA transfection targeting ACA1_031140, ACA1_032330, ACA1_374400, and ACA1_112650, encystation was induced for 48 h. Excystation of Acanthamoeba cysts was observed on non-nutrient agar plates coated with E. coli (A), and the excystation ratio of Acanthamoeba following siRNA treatment was quantified (B). Red arrow, empty cyst. Scale bar=10 μm. Data are expressed as the mean±SD (n=3). **P<0.05, ***P<0.001, and ****P<0.0001.

Table 1Number of genes showing significant increase or decrease (more than 2-fold) during encystation and excystation
Table 1
|
No. of increase |
No. of decrease |
Total |
|
En 8a/Troi
|
1,947 |
1,874 |
3,821 |
|
En 16b/Tro |
2,365 |
2,191 |
4,556 |
|
En 24c/Tro |
2,523 |
2,399 |
4,922 |
|
En 48d/Tro |
2,009 |
1,861 |
3,870 |
|
Ex 8e/Tro |
1,842 |
1,492 |
3,334 |
|
Ex 16f/Tro |
2,182 |
1,718 |
3,900 |
|
Ex 24g/Tro |
2,113 |
1,800 |
3,913 |
|
Ex 48h/Tro |
3,682 |
3,123 |
6,805 |
|
Ex 8/En 48 |
1,214 |
1,163 |
2,377 |
|
Ex 16/En 48 |
1,414 |
1,219 |
2,633 |
|
Ex 24/En 48 |
1,045 |
986 |
2,031 |
|
Ex 48/En 48 |
3,403 |
2,526 |
5,929 |
References
- 1. Siddiqui R, Khan NA. Biology and pathogenesis of Acanthamoeba. Parasit Vectors 2012;5:6.
http://doi.org/10.1186/1756-3305-5-6
- 2. Wang Y, Jiang L, Zhao Y, Ju X, Wang L, et al. Biological characteristics and pathogenicity of Acanthamoeba. Front Microbiol 2023;14:1147077.
http://doi.org/10.3389/fmicb.2023.1147077
- 3. Khan NA.
Acanthamoeba: biology and increasing importance in human health. FEMS Microbiol Rev 2006;30(4):564-595.
http://doi.org/10.1111/j.1574-6976.2006.00023.x
- 4. Coulon C, Collignon A, McDonnell G, Thomas V. Resistance of Acanthamoeba cysts to disinfection treatments used in health care settings. J Clin Microbiol 2010;48(8):2689-2697.
http://doi.org/10.1128/JCM.00309-10
- 5. Mazur T, Hadaś E, Iwanicka I. The duration of the cyst stage and the viability and virulence of Acanthamoeba isolates. Trop Med Parasitol 1995;46(2):106-108.
- 6. Anwar A, Khan NA, Siddiqui R.
Combating Acanthamoeba spp. cysts: what are the options? Parasit Vectors 2018;11(1):26.
http://doi.org/10.1186/s13071-017-2572-z
- 7. Lakhundi S, Khan NA, Siddiqui R. The effect of environmental and physiological conditions on excystation of Acanthamoeba castellanii belonging to the T4 genotype. Parasitol Res 2014;113(8):2809-2816.
http://doi.org/10.1007/s00436-014-3941-6
- 8. Chambers JA, Thompson JE. A scanning electron microscopic study of the excystment process of Acanthamoeba castellanii. Exp Cell Res 1972;73(2):415-421.
http://doi.org/10.1016/0014-4827(72)90066-3
- 9. Li XY, Pi YN, Chen Y, Zhu Q, Xia BR. Nicotinamide N-Methyltransferase: a promising biomarker and target for human cancer therapy. Front Oncol 2022;12:894744.
http://doi.org/10.3389/fonc.2022.894744
- 10. Wang W, Yang C, Wang T, Deng H. Complex roles of nicotinamide N-methyltransferase in cancer progression. Cell Death Dis 2022;13(3):267.
http://doi.org/10.1038/s41419-022-04713-z
- 11. Lorenzo-Morales J, Kliescikova J, Martinez-Carretero E, De Pablos LM, Profotova B, et al. Glycogen phosphorylase in Acanthamoeba spp.: determining the role of the enzyme during the encystment process using RNA interference. Eukaryot Cell 2008;7(3):509-517.
http://doi.org/10.1128/EC.00316-07
- 12. Mattar FE, Byers TJ. Morphological changes and the requirements for macromolecule synthesis during excystment of Acanthamoeba castellanii. J Cell Biol 1971;49(2):507-519.
http://doi.org/10.1083/jcb.49.2.507

 , Hye-Jeong Jo1,†
, Hye-Jeong Jo1,†