Emerging Plasmodium falciparum K13 gene mutation to artemisinin-based combination therapies and partner drugs among malaria-infected population in sub-Saharan Africa
Article information

Abstract
The continuous Plasmodium falciparum kelch13 (PfK13) genetic alterations conferring resistance to artemisinin-based combination therapies and partner drugs pose a significant threat to effective treatment and control of P. falciparum infection in developing countries. This review evaluates the emergence and epidemiology of the PfK13 mutation associated with artemisinin resistance in the sub-Saharan Africa (SSA) population. Despite empirical control and artemisinin combination therapy, the PfK13 gene mutation, previously described in Southeast Asia, has been reported in the SSA. Eight of these validated markers, including P553L, M476I, C580Y, A675V, P574L, R561H, R622I, and F446I, were reported among the SSA population. Novel and unvalidated markers, such as P615S, M472I, F434S, A578S, P570L, Y558C, K563R, A569T, I684N, M472I, and C473F spread among the population with low frequency. We provide insight into the emergence and spread of validated and unvalidated PfK13 mutations among the SSA population, which could lead to high artemisinin resistance. Investigating the verified PfK13 mutations will improve prophylactic strategies, prognostic diagnosis and guide effective population-based surveillance for effective P. falciparum malaria control in SSA.
Introduction
Malaria is one of the most severe public health cases in sub-Saharan Africa (SSA), with 94% global morbidity and mortality rates, mostly among children and pregnant women [1]. Plasmodium falciparum infection is the most severe malaria parasite with high resistance to artemisinin-based combination therapies (ACTs) [2]. Of 18% to 83% of falciparum-infected children in endemic regions of SSA countries [3], high malaria infection rates were reported in Nigeria, Congo, Uganda, Mozambique, Angola, and Burkina Faso [4]. Despite the therapeutic effectiveness of ACTs, the genetic resistance of P. falciparum kelch 13 (PfK13) reported in Southeast Asia has now been found in the SSA population [5]. Delays in the clearance of parasites after therapy with an artemisinin drug indicate partial artemisinin resistance (ART). In vitro and in vivo studies have shown that this delayed clearance of parasites is associated with mutations in the propeller domains of PfK13 and BTB/POZ [2]. With the endemicity of P. falciparum malaria, PfK13 has been identified in SSA populations, which has become an important marker for molecular surveillance [6]. The emergence of the PfK13 mutation calls for urgent preventive measures against potential local PfART-resistance emergence and possible spread in SSA. An intensive investigation for PfK13 markers is urgently required for effective PfART-R monitoring and therapy.
Selection Criteria
A literature search was conducted in the Google Scholar database and PubMed to select peer-reviewed and published studies on pfk13 gene polymorphisms in P. falciparum in the last 10 years, ranging from June 2014 to July 2024. Reported studies from Africa were majorly examined, while few studies from Asia, America, and Europe were included to guide the pre-existence of the PfK13 mutation. Among the search terms used are “Plasmodium falciparum k13 gene,” “artemisinin resistance,” “kelch-13,” “PfK13 gene mutation,” “pfk13-propeller gene,” and “PfK13 marker.” English was used for all searches.
Eligibility criteria for article selection
The included study papers examined the frequency of PfK13 mutations in SSA. Included were publications that examined the pfk13 gene for molecular indicators for ART or that confirmed ART both at the molecular and phenotypic levels. Publications reporting PfK13 polymorphisms sequencing were also included in the selection for evaluation.
Exclusion criteria for ineligible articles
Unrelated studies examining mathematical modeling of resistance, malaria epidemiological studies, and additional Plasmodium-resistant genes were excluded.
Key Findings
For this review, pfk13 gene mutations were reported in 15 studies conducted in the last 10 years (2014 to 2024), mainly in the African population, and a few in Asia, America, and Europe were selected. Generic publications on malaria resistance gene markers other than PfK13 were excluded from the review, while published studies reported in PubMed (n=3) and Google Scholar (n=12) domains were included for the evaluation.
Epidemiology of P. falciparum infection
In 2018, there were approximately 228 million malaria cases worldwide. People with little or no effective immunity are exposed to severe malaria [7]. Severe malaria mainly affects children under 5 years of age in countries, where P. falciparum is persistent and highly contagious with repeated infections. However, people who have a low immune response to P. falciparum infection and who visit endemic areas are at risk for severe malaria at any age due to low or unstable transmission rates [8]. Reduced immune status in pregnant women makes them more vulnerable to malarial infections [9]. While age and gravidity pose a level of risk, young primigravidae in high-transmission areas are most at risk of falciparum infection, mainly in the second trimester [10]. Because of reduced immune responses to malaria infection, Pregnant women who live in regions with inconsistent and low rates of malaria transmission frequently contract the disease quickly, leading to severe illness and mortality [9]. Approximately 85% of the malaria cases worldwide in 2018 were reported from 19 SSA nations as well as India. P. falciparum, which accounts for 99.7% of malaria cases, and is the most severe malaria parasite that is frequently linked to severe anemia, fever, thrombocytopenia, seizures, respiratory distress, metabolic acidosis, hyperglycemia and occasional increased intracranial pressure leading to coma and mortality, particularly in the SSA [11]. Due to the increasing rates of falciparum infectivity with severe clinical pathologies, there are increasing rates of antimalarial drugs, particularly the artemisinin derivatives, that have given rise to prevalent resistance. The misuse, poor prescription, and consumption of adulterated or fake products in several SSA communities intensify mutational changes, leading to high levels of P. falciparum resistance [12].
Artemisinin resistance
P. falciparum-related malaria is often treated with a class of artemisinin and its semi-synthetic derivatives. It comes from Artemisia annua, which is used in traditional Chinese medicine and is known as Sweet Wormwood [13]. Genetically engineered yeasts were substantially more prolific than plants as precursor compounds, with sesquiterpene lactones and a unique peroxide bridge to make up artemisinin and its derivatives [14]. Because P. falciparum requires numerous simultaneous changes to develop drug resistance, ACTs aim to increase the barrier to the erythrocytic proliferation of the parasite within 1 to 3 hours after drug administration. The growing resistance to currently available drugs against malaria is impeding the ongoing global effort to reduce malaria. Even after the drug is absorbed to acceptable concentrations, the ability of Plasmodium parasite strains to endure or multiply continues to cause resistance [12]. Sulfadoxine-pyrimethamine (PND)- and chloroquine-resistant malaria parasites originated following similar pathways, spreading from Southeast Asia through Africa and westward [12]. The cornerstone treatment for malaria mainly consists of ACTs, which combine a partner drug with a longer half-life with an artemisinin derivative’s fast, powerful effect. However, the resistance to artemisinin and its derivatives expanded throughout Southeast Asia, with reports of artemisinin-resistant parasites [15]. Continuous surveillance is required to safeguard artemisinin efficacy and companion drugs in developing countries, as ACTs are currently the preferred, universally effective therapeutic option [16]. Treatment efficacy, clinical investigations, in vitro or ex vivo drug studies, and the evaluation of genetic markers are useful approaches to monitor the emergence and dissemination of drug-resistant P. falciparum [17]. Molecular indicators are utilized more frequently for real-time resistance monitoring, though treatment efficacy studies provide precise information on therapeutic failure [17]. These molecular indicators are markers with differences in copy number or mutational genes encoded in the parasite genome presenting the resistance status of the parasite.
Classes of ACTs
P. falciparum multidrug resistance 1 (pfmdr1) and P. falciparum chloroquine resistance transporter (pfcrt) genes were found to be associated with resistance markers to amodiaquine (AQ), lumefantrine (LMF), and mefloquine (MQ), which are the ACT companion drugs [18]. Compared to pfmdr1 in the wild-type, infection with P. falciparum having the pfmdr1 Asn86Tyr mutation is linked to a higher rate of AQ treatment failure [19]. Infection with parasites lacking the mutation is linked to a lower rate of artemether-LMF treatment failure. In endemic areas of SSA, single or combined first-line therapy (usually artemether-LMF or artesunate (AS)-AQ) is used for uncomplicated malaria. The World Health Organization (WHO) suggests switching to first-line treatment when clinical resistance prevalence rises above 10% [20]. Artemisinin molecular markers serve as surveillance tools for assessing and monitoring drug resistance among the population. It is useful for tracking observable shifts in the drug resistance landscape [17]. A decreasing prevalence of resistance marker alleles indicates a low susceptibility. A shift in national policies regarding children’s intermittent preventative treatment has been justified by the frequency of mutations associated with sulfadoxine-PND resistance [7]. The WHO recommends AS-pyronaridine, dihydroartemisinin-piperaquine (DHA-PPQ), AS-MQ, AS-sulfadoxine-PND AS-AQ, artemether-LMF for malaria treatment (Table 1) [2]. ACTs’ introduction has facilitated drastically minimization of malaria cases and lowered mortality rates [2]. The following section explains the classes of artemisinin partner drugs currently under threat due to the emergence of pfk13 gene mutation.
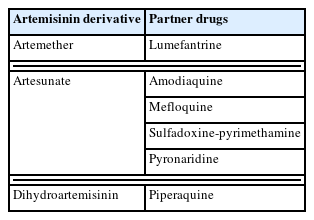
Resistance to ACTs
As ART resistance persists, it is crucial to keep an eye on sustaining the effectiveness of the ACT companion drugs. Drug recycling is necessary given our limited supply of antimalarials; nevertheless, this should only be done in cases when resistance to both current and prior drugs is well-established. The most utilized companion drugs include antifolates (sulfadoxine-PND), arylamino alcohols (LMF and MQ), and aminoquinoline medicines (AQ, PPQ, and PND). In Southeast Asia, MQ and PPQ are the most utilized companion drugs, but LMF and AQ are widely used in Africa. A recent study of companion drug resistance pathways provided an overview of companion drug resistance in the context of ACT resistance, with a particular emphasis on LMF, MQ, AQ, and PPQ [21,22].
PPQ
When AS-AQ failure rates rose too high in the last 10 years, DHA-PPQ were replaced in Cambodia. DHA-PPQ was utilized in Asia and Africa for mass drug administration purposes [23] and as a routine therapy for malaria during pregnancy [24]. However, as ART resistance began to arise, clinical resistance to DHA-PPQ was also observed in Vietnam and Cambodia [25,26]. The situation quickly worsened in the eastern Greater Mekong sub-region, underscoring the urgent need for effective interventions. These findings have significant implications for the future of malaria treatment and drug resistance and underscore the importance of continued research in this field.
Studies on the 2-food vacuole membrane resident transporters, PfMDR1 and PfCRT, prompted the effect of PPQ in the food vacuole. A novel PfCRT mutation (C101F) was found in parasites with in vitro PPQ resistance [27]. The expression of C350R mutation in PfCRT was linked to increased PPQ IC50 values, according to a report of field isolates from French Guiana [28]. Three unique PfCRT mutations— M343L, G353V, or H97Y—that were linked to high PSA readings were found in re-emerging parasites treated with DHA-PPQ. Furthermore, Genome-wide association studies linked the PfCRT F145I mutation to reduce ex vivo PPQ sensitivity and treatment failure for DHA-PPQ [29]. Genetic validation has confirmed several PfCRT mutations as PPQ resistance indicators. Crucially, PPQ susceptibility was considerably decreased by the development of certain mutations, G353V, F145I, and M343L, regardless of plasmepsin 2 or 3 amplification [28]. These genomic amplifications may, therefore, contribute to establishing a favorable genetic background that gave rise to pfcrt mutations or serve as a baseline for low-grade PPQ resistance [30]. In conjunction with plasmepsin amplification, drug pressure, and the corresponding fitness costs dictate the emergence and spread of these PfCRT haplotypes. After the introduction of DHA-PPQ, there was a noticeable rise in the prevalence of the novel PfCRT mutations, as shown by longitudinal surveillance of parasites from Cambodia, with >98% of parasites bearing these changes by 2017 [31]. PfCRT’s structural insights have clarified how important it is for preventing drug resistance to malaria. The wild-type protein at these 2 locations can only actively efflux chloroquine (CQ) but not PPQ, leading to CQ resistance. In contrast, the 7G8 isoform of PfCRT with C350R and F145I mutations binds and transports PPQ out of the food vacuole in membrane potential- and pH-dependent ways, resulting in PPQ resistance [32]. This leads to the preferential binding of specific PfCRT isoforms to release CQ or PPQ, which regulates drug resistance to these drugs and adds to collateral drug sensitivity [32].
AQ
As an adjunct to treating uncomplicated P. falciparum malaria, AS-AQ is the second most used ACT in Africa for seasonal malaria chemoprevention [33]. Like CQ, the main factor determining AQ resistance is a mutation in PfCRT, with PfMDR1 alterations having supporting functions [34]. AQ resistance is specifically linked to a PfCRT haplotype of SVMNT. The PfCRT structure could account for the parasites’ varying susceptibility to CQ and AQ. AS-AQ similarly selects for the AQ resistance-correlated mutant PfMDR1 86Y, similar to CQ [35].
Mefloquine and LMF
In Thailand, AS-MQ eventually replaced MQ, initially used with sulfadoxine-PND and then as a monotherapy [36]. Although indications of reduced efficacy have been recorded in several African countries, artemether-LMF is the most often used ACT in the continent [37]. This combination was utilized in the eastern Greater Mekong sub-region in 1990 to overcome the resistance associated with MQ monotherapy. The lack of strong data has sparked disagreement about recent research indicating low efficacy for artemether-LMF in Angola [38]. Although the exact mechanism of action of these arylamino alcohols is unknown, it is anticipated that they will obstruct heme detoxification, albeit to a lesser extent than CQ [39]. These medications may also impair PfMDR1’s ability to operate normally. According to a recent study, MQ binds to the large ribosomal subunit’s GTPase-associated core [40]. Clinical research has shown, as predicted, that DHA-PPQ selects for the mutant allele, whereas artemether-LMF selects for wild-type PfMDR1 at N86 [40]. Triple ACTs are now being designed using the theory that the selection for resistance to one treatment results in hypersensitivity to another drug.
Mechanism of resistance of P. falciparum to artemisinin
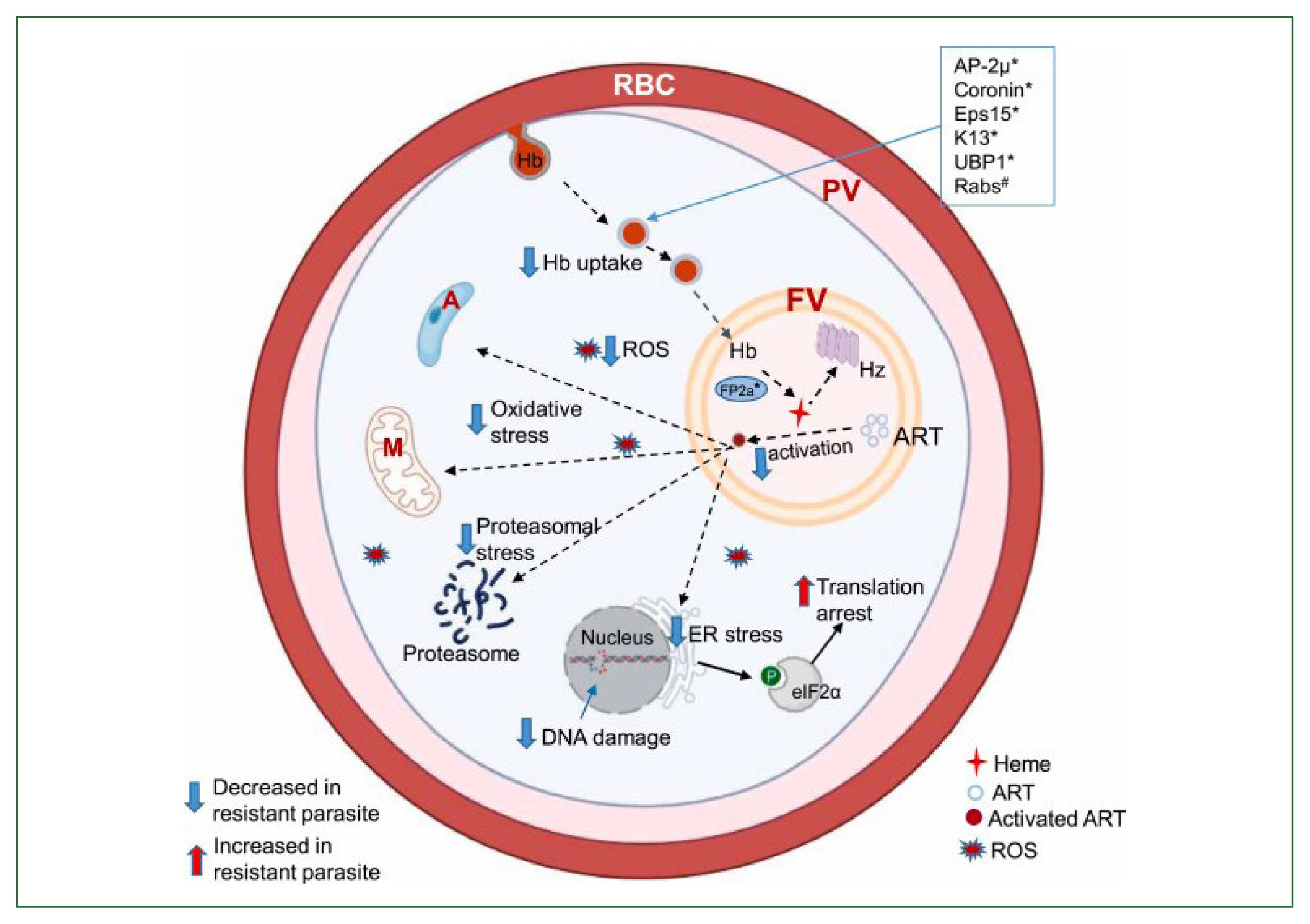
Proteins such as AP-2, Coronin, Eps15, K13, UBP1, and Rab GTPases, among others, are thought to be involved in the endocytosis process. Hemoglobin is broken down by numerous hemoglobinases inside the food vacuole, most notably Falcipain 2a, which releases toxic heme for the parasite. The inactive form, hemozoin, is then created from heme. Heme is additionally required for the activation of ART, though. Eukaryotic initiation factor 2 is phosphorylated, and an oxidative stress response is triggered when ART is engaged, among other cellular targets. Reactive oxygen species are also released by the parasite’s cytoplasm and mitochondria, which can harm the parasite’s DNA (Fig. 1) [36]. The heme synthesis is lowered, ART is activated less, and cellular stress is lessened in the ART-resistant parasite due to altered protein variants linked to decreased hemoglobin uptake and digestion. When K13 was conditionally inactivated by mislocalization to the nucleus (knock sideways), ring-stage survival assay ART susceptibility was decreased, and the number of hemoglobin-like peptides was increased, indicating a decrease in hemoglobin catabolism. This made the link between endocytosis and K13-mediated ART resistance evident [41]. It has been discovered that field isolates that are resistant to culture also have impaired hemoglobin digestion [42]. Young parasite rings with the K13 mutation confer resistance and endocytose less inside the host cell cytosol, demonstrating a connection between susceptibility and endocytosis. Earlier research has proved that preventing hemoglobin from being digested or conditionally inactivating the P. falciparum food vacuole protease reduced vulnerability to ART [43]. Following hemoglobin breakdown, heme is released and can initiate ART. The downregulation of the Heme Crystal-Catalyzing Unit results in less ART activation and a lower concentration of the active drug, which follows K13’s function in hemoglobin endocytosis and resistance [43]. The K13-propeller mutation increases the concentration of phosphatidylinositol-3-phosphate by preventing uridine triphosphate fluctuation (uTF) and PI3K from adhering to the Kelch domain before being ubiquitinated. It was reported that the buildup of incorrectly folded proteins in the endoplasmic reticulum is caused by oxidative stress brought on by artemisinin [44]. When misfolded proteins link to BiP, PfPK4 disassembles the complex BiP-PfPK4 and phosphorylates uTF and eukaryotic translation initiation factor 2. According to [44]. Eukaryotic translation initiation factor 2 phosphorylation should enable uTF translocation in the nucleus to control the expression of cytoprotective genes and unfolded protein response targets while repressing protein synthesis (Fig. 2) [44].
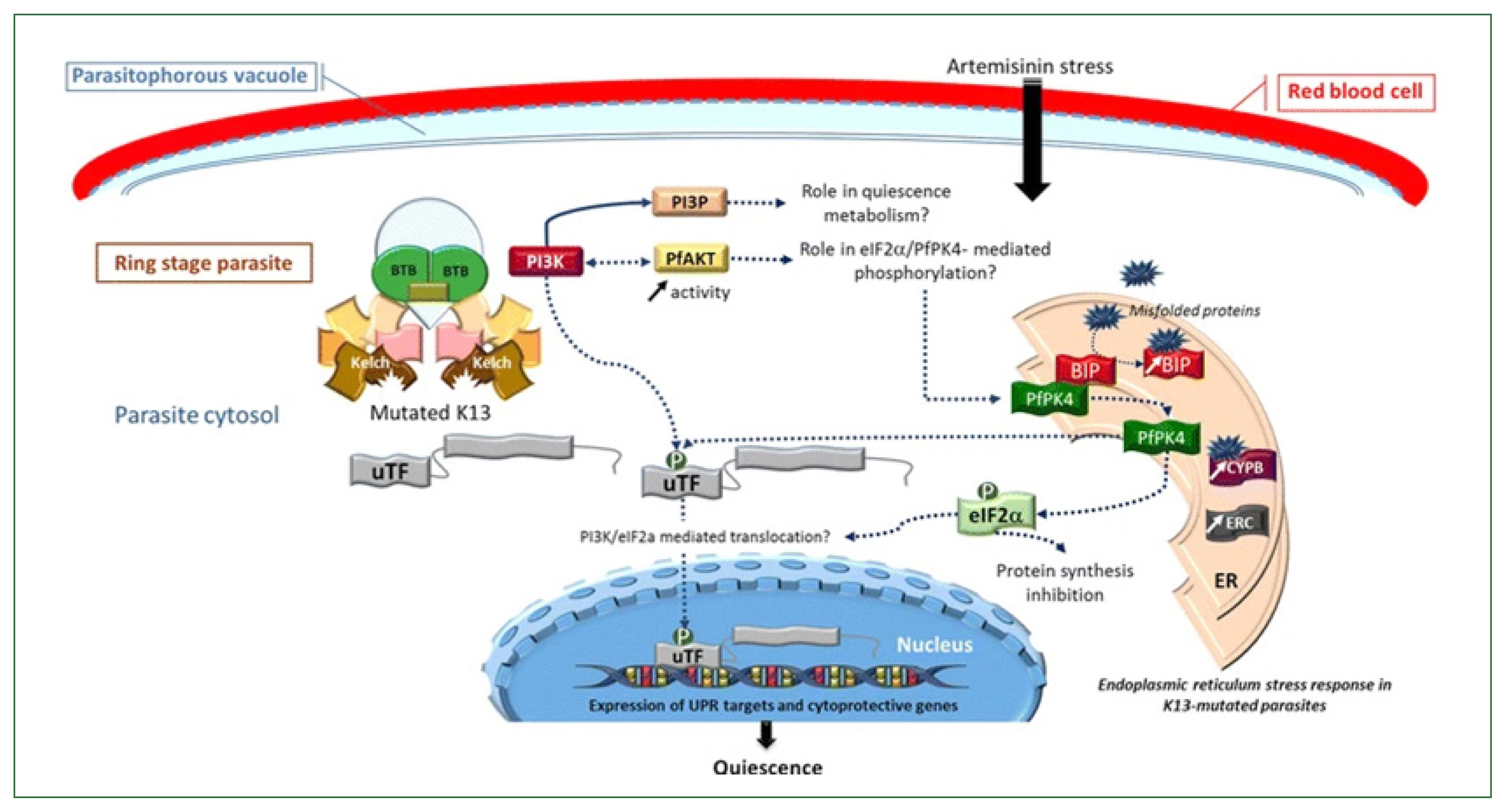
Significance of the K13 gene mutation in P. falciparum
The K13 protein, with a sequence comparable to the Kelch/BTB/POZ homogeneous adapter family and its 726 amino acids, plays a pivotal role in the degradation process. When a protein is no longer required, polyubiquitination, a post-translational alteration, marks it for proteasome degradation. The additional fates of proteins that can be indicated by polyubiquitination include changes in enzyme activity, intracellular traffic signals, and interactions between proteins [45]. The Cullin-3 E3 ubiquitin ligase family collaborates with the Kelch/BTB/POZ adaptor family to supply ubiquitins to regulatory proteins (typically) [46]. This regulation includes cell cycle, gene expression, membrane transport, and endocytosis. Different organisms’ BTB/POZdomains (Linge Fingers of the Bitric-a-brac/Pox virus) have sites that bind the adapter to Cullin-3 and encourage homodimerization [47]. Six Kelch motifs, together forming a 6-bladed propeller structure and denoting “chalice” (chalice) in Flemish, are found in the C-terminal domain. Substrates are anticipated to be engaged by this shallow cup-shaped domain and delivered for ubiquitination. The 2 common sites for changes that cause resistance to malaria treatment are the kelch domain of K13’s surface and the propeller-like core of P. falciparum The mutations may change how K13 interacts with the substrate being given for ubiquitination or cause the structure of K13 to become unstable [48]. Some parasites can quickly reverse the effects of the artemisinin treatment, indicating that they resist the drug. As a result, mutations in the K13 gene were recognized as an indicator of resistance to artemisinin [49].
Origin of K13 gene mutation
Early examples of ART were first observed in Cambodia, characterized by P. falciparum parasites that took a long time to disappear after ART treatment. It was shown that single-point mutations in the Kelch 13 were the primary contributor to resistance [50]. The WHO has categorized about 20 of these changes as validated or candidate markers of ART resistance due to their connection with delayed parasite elimination when assessed in medicine (persistence on day 3 or clearance half-life >5 h) or in vitro (asexual parasite survival >1% in the ring stage assay) [2]. Over 2 hundred non-synonymous variants of the k13 gene have been found in populations of parasites worldwide [51].
Prevalence of K13 gene mutation in SSA
Despite the rarity of K13 mutations in malaria parasites worldwide, it is unknown if K13 mutations found beyond the Greater Mekong sub-region are related to clinical or in vitro delayed clearance features. Recent studies undertaken in Uganda and Rwanda have sparked concerns regarding the emergence of ART resistance in Africa [52–54]. Table 2 gives the summary of the validated Plasmodium falciparum k13 markers reported in SSA countries [1,6,52,55–61]. The K13 R561H mutation was first identified in Rwanda in 2014, and isolates taken from adjacent African regions in 2018–2019 with a prevalence of 20%–22% were also found to have the mutation [52,53]. In patients receiving ART, this mutation has also been linked to a delay in parasite clearance [52,53]. When inserted into Asian or African strains via CRISPRCas9 editing, R561H was found to confer a modest level of ART resistance. This specific mutation was originally identified in Rwanda by de novo evolution [52]. A genetic change (M476I) was discovered in Tanzanian parasite variants after almost 5 years of intermittent ART medication treatment [6]. An extensive number of mutations were centered in the “propeller” region of the K13 protein (Fig. 3) [62], according to a retrospective examination of parasite samples from Cambodia. The longer PC1/2 phenotype and higher in vitro ring stage assay values are substantially linked with this observation, indicating that the k13 gene plays a key role in determining the delay parasite clearance phenotype. In investigations on clinical efficacy, several K13 mutations have been connected to delay parasite clearance [5]. Numerous common K13 polymorphisms (R539T, N458Y, C580Y, Y493H, and I543T) were discovered during in vitro ART-resistant testing by genetic analysis [62], offering an indicator gene for ART resistance surveillance.
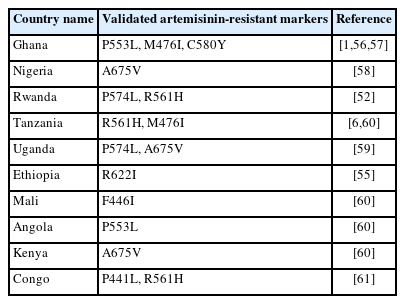
List of validated K13 markers in sub-Sahara Africa
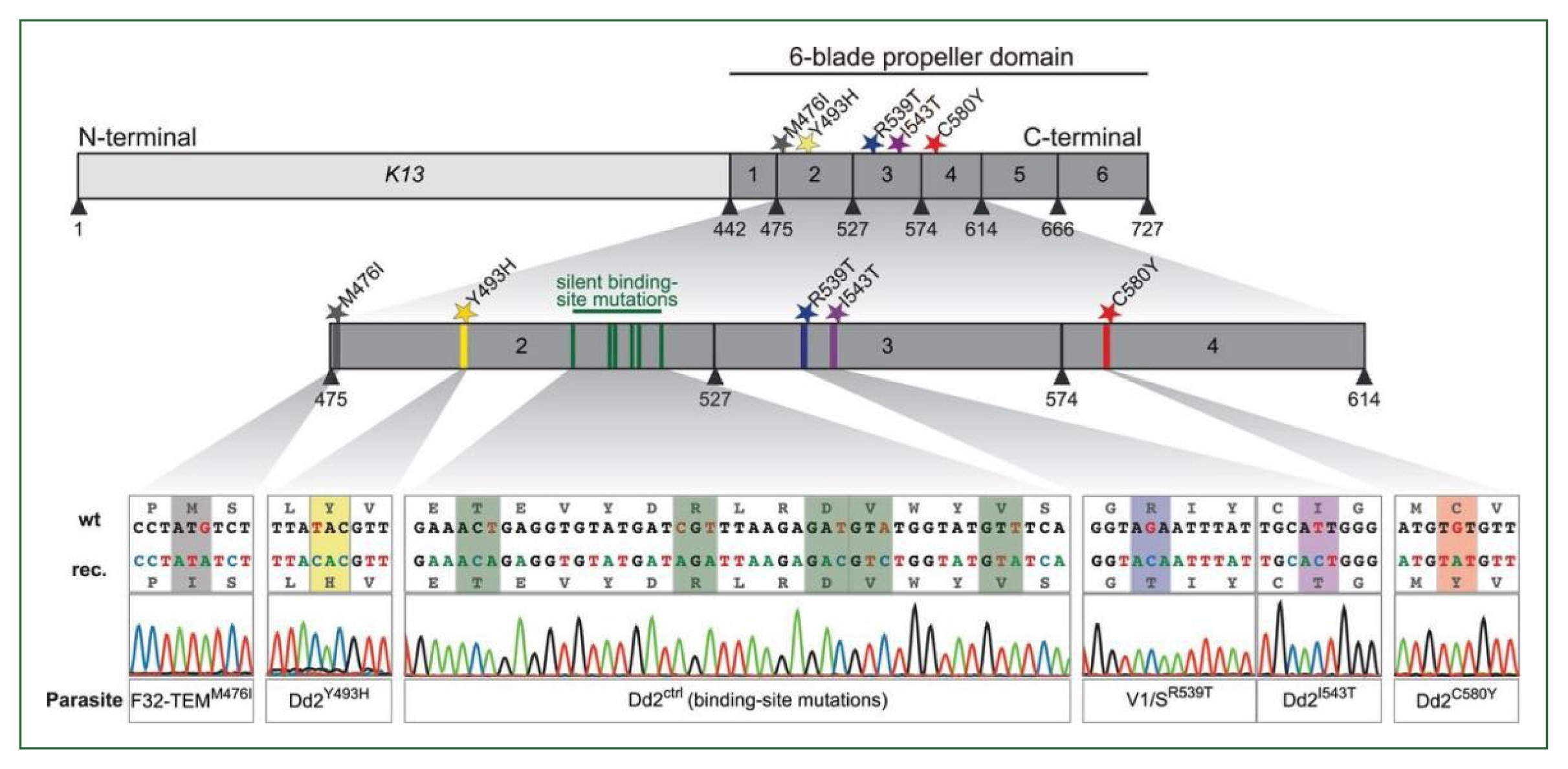
There are more K13 polymorphisms in African parasite samples than in the Greater Mekong sub-region but fewer delay parasite clearance-related modifications [36]. Molecular surveillance studies showed several single nucleotide polymorphisms (SNPs) have been found in African nations, such as P570L, Y558C, K563R, and P615S M472I in Nigeria [63], R622I in Ethiopia [55], C473F in Senegal [1] and F434S, I684N was discovered in Nigeria in addition to M472I and A569T in Congo [1]. The most prevalent mutation in SSA countries, including Nigeria, Gabon, Ghana, Kenya, the Congo, Uganda, Cameroon, and Mali, is the A578S SNP. The PfK13 protein’s structure may change due to this mutation, which swaps out a non-polar amino acid for a polar one. More focus must be placed on it because SSA is highly prevalent in the pfk13 gene [64]. These discovered SNPs might contribute to ART resistance even if their functional roles have not yet been established. These, however, should be taken into consideration as drug pressure increases and ART resistance in Africa rises [64]. The unsettling discovery of resistance-mediating K13 mutations outside of the GMS in multiple recent studies emphasizes the impending potential for ART resistance to spread elsewhere. The increasing incidence of the ART marker warrants concern. In a recent study, 20 of the 124 non-synonymous variants in the K13-propeller were discovered to be “associated” with ART [65]. Some of these variants include G538V, N458Y, C469Y, R539T, F673I, I543T, P553L, C580Y, F446I, P44L, G449A, V568G, D584V, S522C, A481V, Y493H, P574L, A675V, H719N and R561H. R539T, I543T, Y493H, and C580Y are the only 4 of these mutations that are validated markers. The WHO included 9 non-synonymous mutations in PfART-R among the African PfK13 alleles, primarily based on Southeast Asian studies [66]. In vivo parasite development clearance was revealed to be substantially correlated with 6 of these (P553L, P446I, M476I, V568G, P574L, and A675V) [61]. WHO acknowledges P446I, M476I, and P533L as verified ART indicators, nevertheless, because in vitro evidence of diminished response to the drug supports this [67]. Even though it is not included as a validated marker, A675V has demonstrated a unique responsiveness to ART in one diagnostic research from Uganda. P446I was only found in Mali, which is in West Africa, but A675V, M476I, P553L, P574L, and V568G were all observed in the Great Lakes region of East Africa [67]. The WHO states that the 3 additional mutations—S522C, C469T, and A515L—found in African isolates are inconsequential despite the possibility that they may contribute to artemisinin’s slower parasite clearance. The WHO has so far validated thirteen markers for ART: Y493H, M476I, R539T, I543T, F446I, C469Y, N458Y, R561H, C580Y, P574L, R6221, A675V and P553L. Nine other associated markers—P441L, R515K, C469F, N537I/D, A481V, P527H, V568G, G449A, and G538V—are also present [2]. The previously identified mutations R622I, C469Y, and A675V found in various African countries have been proven to be related to ART. According to recent studies from Uganda and Nigeria, samples from those nations gathered in 2018 and 2019 showed a considerable increase in the prevalence of the A675V mutation. For example, the A675V mutation was present in 5% of the Ugandan samples. The A675V mutation was also noticed in samples from Nigeria and Rwanda [56]. The A675V mutation has been validated for the ART-R marker in Kenya [56], Ethiopia possesses the R622I mutation [55], P574L was found in Rwanda [52], Ghana has P553L [56]; whereas Uganda has the C469Y mutation [53]. Malaria eradication remains elusive worldwide, with transmission persisting in numerous endemic nations [68]. P. falciparum parasites are resistant to all known malaria drugs, and their ongoing emergence is a key obstacle to malaria elimination attempts [69,70]. C580Y has also been reported in Ghana [1,57], A675V in Nigeria [58], P574L and A675V in Uganda [59], R561H in Tanzania, F446I in Mali, P553L in Angola, and A675V in Kenya [60]. Additionally, P441L and R561H have been reported in the Congo [61]. This is a concern because, according to migration patterns, these mutated malaria parasites would likely spread rapidly to neighboring African nations within a short period. To prevent dissemination, appropriate control measures are needed to prevent resistance development and failure to treatment. It is essential to routinely monitor the spread as a result of the occurrence of these mutations in the pfk13 gene.
Conclusion
This evaluation calls for preventive measures against potential local PfART-resistance emergence and possible spread in sub-Sahara Africa. An intensive investigation for PfK13 markers is urgently required for effective PfART-R monitoring and therapy. To determine their potential roles in establishing and disseminating ART-decreased susceptibility/resistance among children in SSA, in vivo and in vitro research must be conducted to prevent the impending emergence of mutated Pfk13 strains among the African population.
Notes
Author contributions
Conceptualization: Oyegbade SA, Akinduti PA
Data curation: Oyegbade SA, Akinduti PA
Formal analysis: Oyegbade SA, Mameh EO, Akinduti PA
Funding acquisition: Balogun DO, Aririguzoh VO, Akinduti PA
Investigation: Oyegbade SA, Mameh EO
Methodology: Oyegbade SA, Akinduti PA
Project administration: Oyegbade SA, Mameh EO, Balogun DO, Aririguzoh VO,
Resources: Mameh EO, Balogun DO, Aririguzoh VO, Akinduti PA
Software: Oyegbade SA, Balogun DO, Akinduti PA
Supervision: Oyegbade SA, Akinduti PA
Validation: Oyegbade SA, Akinduti PA
Visualization: Oyegbade SA, Mameh EO, Akinduti PA
Writing – original draft: Oyegbade SA, Mameh EO, Balogun DO, Aririguzoh VO
Writing – review & editing: Oyegbade SA, Mameh EO, Akinduti PA
Conflict of interest
The authors declare no conflict of interest related to this study.
Acknowledgments
The authors thank the management of CUCRID, Covenant University, Ota, Nigeria for providing the publication funding support and Covenant Applied Informatics and Communication Africa Center of Excellence (CApIC-ACE) support in data collection.