Possible association between Toxoplasma gondii infection and autism spectrum disorder
Article information

Abstract
Toxoplasma gondii is a neurotropic apicomplexan protozoan estimated to affect approximately 30% of the global population. In this review, we aimed to examine scientific evidence on the potential role of T. gondii infection in the development of autism spectrum disorder (ASD), a heterogeneous neurodevelopmental disorder. This review summarizes the current literature exploring the possible association between T. gondii and ASD. Findings indicate that toxoplasmosis may contribute to host alterations, including the induction of humoral and cellular immune responses, production of various cytokines, and changes in neurotransmitter levels (e.g., serotonin, dopamine, acetylcholine, gamma-aminobutyric acid, and glutamate), as well as the activation of enzymes such as indoleamine 2,3-dioxygenase, which may influence the pathophysiology of ASD. In conclusion, this review suggests that T. gondii infection could act as a potential risk factor for ASD. However, further intensive studies are necessary to clarify the role of this parasite in the etiology and progression of ASD. This review is anticipated to stimulate further studies aimed at understanding and potentially reducing the burden of neurodevelopmental disorders worldwide.
Introduction
Autism spectrum disorder (ASD) is a heterogeneous neurodevelopmental disorder characterized by impairments in social interaction and communication as well as by the presence of unusual repetitive behaviors that typically emerge in early childhood [1,2]. According to the Autism and Developmental Disabilities Monitoring Network of the US Centers for Disease Control and Prevention, the estimated prevalence of ASD among 8-year-old children in 2020 was 1 in 36—approximately 4% in boys and 1% in girls [3]. ASD has a multifactorial etiology involving neurological, environmental, immunological, and genetic factors, although the precise role of each factor remains unclear [4]. These uncertainties have limited the development of effective treatments. Therefore, improving our understanding of the underlying pathology of ASD is essential for identifying new therapeutic approaches for this lifelong condition [2].
Toxoplasma gondii is an obligate intracellular parasite estimated to infect approximately 25%–30% of individuals in both developed and developing countries [5–7]. It invades various host cell types and forms cysts that usually remain dormant throughout the host’s lifetime [8]. These cysts mostly develop in the brain cells—particularly neurons, astrocytes, and microglia [9]—as well as in the retina and muscle tissues [10]. Emerging evidence suggests that latent toxoplasmosis is not only asymptomatic but can also be harmful [11]. Cysts of T. gondii have been shown to alter neuronal biological functions, including gene expression and neurotransmitter synthesis, potentially disrupting brain connectivity and contributing to behavioral and neurological impairments [12–18]. Recent studies have identified toxoplasmosis as a potential risk factor for several neurodegenerative and neuropsychiatric disorders, including Parkinson disease, Alzheimer disease, schizophrenia, ASD, bipolar disorder, obsessive-compulsive disorder, depression, and suicidality [8,12,19–27]. However, the pathogenic mechanisms underlying these associations remain unclear. Therefore, this review was designed to examine the potential association between toxoplasmosis and ASD. The risk factors through which T. gondii infection may influence this relationship are outlined below, and Table 1 summarizes the key literature supporting these associations [28–39].
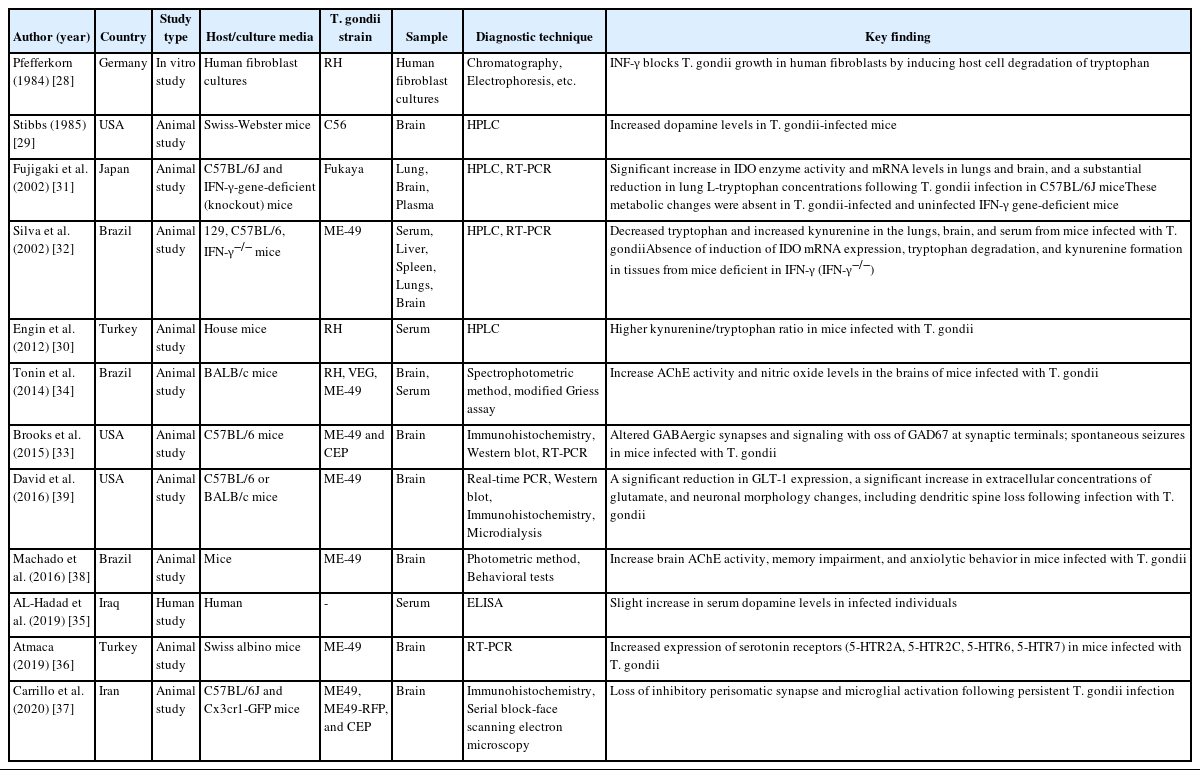
Key literature supporting the role of Toxoplasma gondii infection as a risk factor for autism spectrum disorder
Immunological Factors
The development of neuropsychiatric disorders associated with T. gondii is likely mediated by both the host’s immune response to the parasite and the parasite’s biochemical activity. During T. gondii infection, components of the innate immune system trigger an adaptive immune response by processing antigens and producing cytokines. Microglia, resident macrophages of the central nervous system (CNS), are believed to be the primary cells responsible for limiting the proliferation of T. gondii in the brain, largely through mechanisms mediated by interferon-gamma (IFN-γ). IFN-γ is a critical cytokine involved in controlling both the acute and chronic stages of toxoplasmosis. Elevated levels of IFN-γ, interleukin (IL)-1, and tumor necrosis factor-alpha (TNF-α) have been observed in the brain following T. gondii infection. Moreover, administration of anti-IFN-γ antibodies has been shown to increase the severity of toxoplasmic encephalitis [40]. IFN-γ also stimulates microglia to secrete TNF-α, thus contributing to a regulatory feedback loop in microglial activation. This IFN-γ-mediated microglia activation, along with TNF-α autocrine signaling, likely plays a key role in the defense mechanism of the brain against T. gondii. Activated microglia can exert direct lethal effects on T. gondii by secreting IL-1, TNF-α, and inducible nitric oxide synthase or indirectly by interacting with CD4+ and CD8+ T cells to produce additional cytokines, such as IL-10, IL-12, and IL-15 [40].
Immune dysfunction is considered a potential contributing factor to the development of ASD. Several studies have reported the dysfunction of T cells, the presence of autoantibodies in peripheral blood, and elevated levels of proinflammatory cytokines, chemokines, and differentiation factors in patients with ASD [8,41].
The populations of CD4+ and CD8+ lymphocytes are reduced in children with ASD. An imbalance in T helper 1 and T helper 2 cytokines has also been reported, with a shift favoring the T helper 2 response [25]. In children with ASD, serum immunoglobulin levels are altered due to a significant increase in total protein and serum concentrations of IgG, IgG2, and IgG4, which may be associated with greater susceptibility to infections [42]. These alterations in immunoglobulin classes and subclasses represent one mechanism through which the immune system may contribute to the pathogenesis of ASD [43]. Given that immunoglobulin levels are naturally low at birth and it takes years to reach adult levels—particularly for some isotypes—they are especially relevant in pediatric disorders [44]. Several studies examining immunoglobulin profiles in patients with ASD have found decreased serum IgA [45], elevated plasma concentrations of IgM and IgG [42,46], and increased total IgE [47]. If such abnormal immune activity occurs during vulnerable and sensitive periods of neurodevelopment, it may result in neurological impairments characteristic of ASD. Elevated serum IgG levels in individuals with ASD may contribute to underlying autoimmune processes and/or increased infection risk. The binding of IgG to epithelial cell surfaces, infiltration of lymphocytes, and increased cellular proliferation in the small intestines of children with ASD suggest a higher likelihood of autoimmunity in severe cases [46]. A systematic review and meta-analysis reported an overall odds ratio of 1.93 for anti-T. gondii IgG antibodies in patients with ASD compared to controls, indicating a potential association between T. gondii infection and ASD pathogenesis. However, no significant difference was observed in the anti-T. gondii IgM antibody levels between patients with ASD and controls [48]. During T. gondii infection, IgG antibodies typically appear approximately 2 weeks after the onset of IgM, peak within 2–3 months, and then gradually decline to lower levels, remaining detectable for life due to the persistence of cysts in immune-privileged organs [49,50]. Conversely, IgM antibodies serve as indicators of recent infection, appearing about 1-week postinfection, peaking after approximately 1 month, and declining to undetectable levels over the following months [51].
Kynurenine Metabolic Pathway
During T. gondii infection, IFN-γ and other cytokines produced by neurons, glial cells, natural killer cells, and T lymphocytes play an essential role in the formation of neurodegeneration and oxidative stress associated with ASD [25]. Oxidative stress results from a biological imbalance between reactive oxygen species and antioxidants, leading to changes in biomolecules and the disruption of intracellular redox-related signaling pathways [52]. Natural killer cells are activated early in toxoplasmosis by IL-12 and produce high levels of IFN-γ, which helps control the parasite before T-cell activation [53]. IFN-γ inhibits T. gondii growth by inducing the release of indoleamine 2,3-dioxygenase (IDO), which in turn catalyzes the degradation of tryptophan [28]. The depletion of tryptophan inhibits the growth of the tachyzoite form of T. gondii [29].
Tryptophan is degraded by the IDO enzyme into kynurenine, which is further hydroxylated into quinolinic acid, a potent N-methyl-D-aspartate receptor, or metabolized into kynurenic acid, an endogenous antagonist of the glutamate N-methyl-D-aspartate receptor [54]. These metabolites exert neurotoxic effects (quinolinic acid) and propsychotic effects (kynurenic acid), potentially disrupting the neurotransmitter balance (Fig. 1) [55].
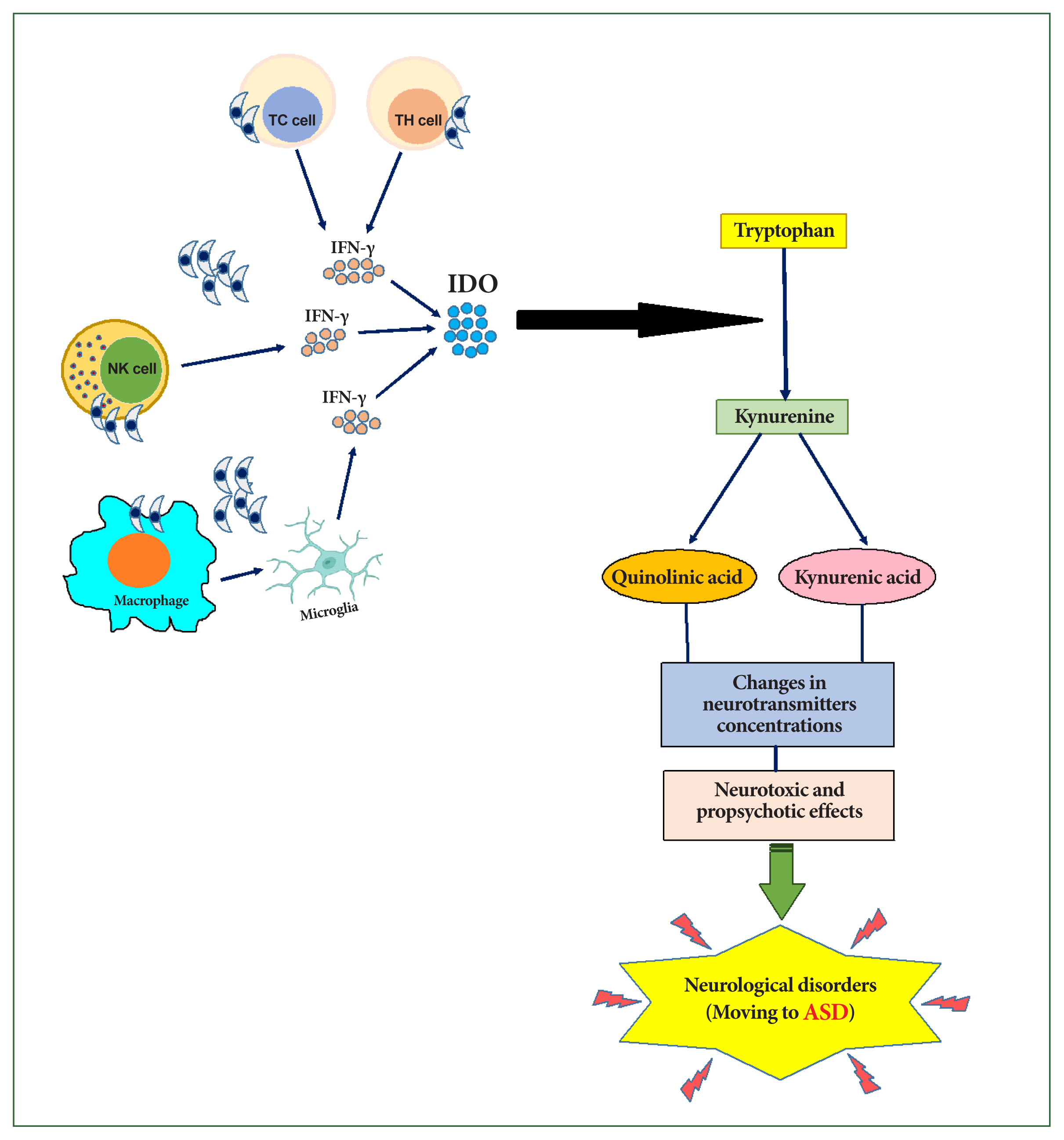
Role of interferon-gamma (IFN-γ) in stimulating the kynurenine metabolic pathway following Toxoplasma gondii infection, contributing to neurological disorders. IDO, indoleamine 2,3-dioxygenase; ASD, autism spectrum disorder.
Stimulation of the kynurenine metabolic pathway following T. gondii infection may represent a host defense mechanism against pathogen infections [56]. However, high levels of kynurenic acid and quinolinic acid have been linked to various neurological disorders. Schwartz [57] suggested that abnormal tryptophan metabolism may serve as a unifying biochemical basis for ASD progression.
Significant decreases in tryptophan levels have been observed in the serum and brain of mice infected with T. gondii [30–32]. Correspondingly, studies have reported substantial increases in kynurenine levels in either serum or brain following toxoplasmosis [31,32]. Maximal changes in both tryptophan and kynurenine levels were detected between days 5 and 20 postinfection [32]. Inflammatory processes thus notably shift the tryptophan pathway toward kynurenine metabolites, disrupting the balance between physiological function and neurotoxicity (Fig. 1).
Given the clinical features of congenital toxoplasmosis, such as intracranial calcifications, hydrocephalus, and intellectual dysfunction [58], T. gondii may be considered a risk factor for ASD. Delayed motor development and intellectual dysfunction are common among children exposed to the parasite in utero, with some remaining asymptomatic until school age [59,60].
This evidence supports a potential link between neuroinflammation, kynurenine, and ASD. Since kynurenine can affect various neurotransmitter systems and is influenced by environmental factors, including infection, stress, and inflammation, it is plausible that kynurenine plays a central role in ASD etiology [61].
Chronic toxoplasmosis induces specific neuropsychiatric symptoms and behavioral changes in humans and animals; however, the mechanisms responsible for these changes are not fully understood. Parasite-induced alterations in CNS neurotransmitters are among the most essential factors implicated in these psychiatric consequences (Fig. 2) [11,33–35].
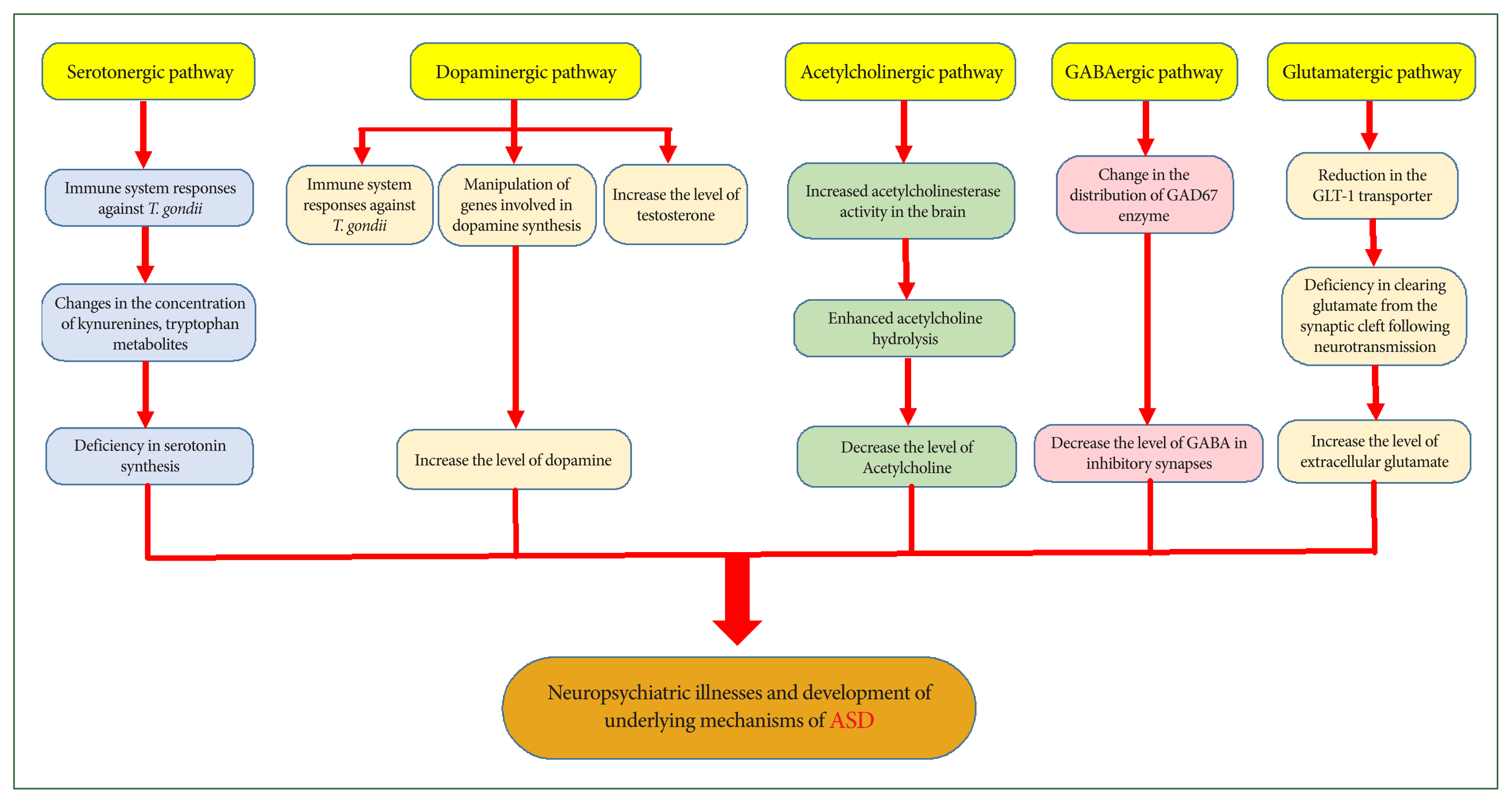
Effects of toxoplasmosis on neurotransmitter pathways in the central nervous system, ultimately leading to neurological disorders. GAD, glutamate decarboxylase; GABA, gamma-aminobutyric acid; GLT-1, glutamate transporter-1; ASD, autism spectrum disorder.
The response of the immune system to T. gondii may alter neurotransmitter concentrations by modifying the concentrations of kynurenines, tryptophan metabolites, and other endogenous brain compounds [62]. Serotonin (5-HT) plays an important neurotrophic role during brain development, and abnormalities in CNS concentration can disrupt its functions. Tryptophan is an essential substrate for serotonin synthesis [63]. Serotonergic abnormalities, along with related cortical development disruptions, are common mechanistic features of ASD. An initial deficiency in serotonin synthesis may affect extensive brain regions, contributing to mood alterations, impaired social interaction, sleep disturbances, aggression, and obsessive-compulsive behaviors reported in ASD [64–66]. Research suggests that serotonin-related psychiatric disorders, such as hyperserotonemia, occur in 25%–50% of individuals with ASD and their first-degree relatives, particularly their parents [67,68].
Furthermore, T. gondii directly and indirectly affects dopaminergic signaling and activates the kynurenine metabolic pathway, diverting tryptophan away from serotonin production [69]. Host immune responses to T. gondii infection induce T helper 1 differentiation and the production of IFN-γ cytokine, which stimulates IDO enzyme activity to inhibit parasite growth. Activation of this enzyme depletes tryptophan reserves, thereby reducing serotonin synthesis in the brain [36]. This reduction may contribute to neuropsychiatric disorders and is particularly critical in cases of congenital toxoplasmosis, as studies have shown that abnormal serotonin levels during prenatal and early postnatal development, together with cortical developmental alterations, may predispose individuals to ASD (Fig. 2) [64].
Dopamine and the Dopaminergic Pathway
Dopamine is a neurotransmitter that plays a central role in reward-motivated behavior and motor control, and its dysfunction has been implicated in various neurological and psychiatric disorders [70]. Among the numerous genes manipulated by T. gondii in an infected host is one that encodes the enzyme tyrosine hydroxylase, which is involved in synthesizing the dopamine neurotransmitter—often referred to as the “pleasure molecule” [35,71]. Evidence suggests that increased testosterone levels during chronic toxoplasmosis may enhance dopamine production and that T. gondii can also synthesize dopamine [37,72]. Nitric oxide, IL-2, and IL-6, which are produced by the host immune response to the parasite, further stimulate dopamine release [72]. Consequently, as shown in both human and animal studies, chronic toxoplasmosis leads to elevated dopamine concentrations in the CNS, and dopamine dysfunction has long been associated with various neuropsychiatric disorders (Fig. 2) [11,35,73,74]. Notably, individuals with ASD often exhibit high dopamine levels, and antipsychotic medications that block dopamine receptors are among the most effective treatments for ASD, supporting a significant role for dopamine in the manifestation of ASD symptoms [65,75].
Acetylcholine
Acetylcholine is a neurotransmitter that may play a key role in the relationship between latent toxoplasmosis and ASD, as alterations in acetylcholine or its receptors have been reported in both conditions and are potentially linked to specific neurological and behavioral symptoms. Acetylcholine affects the brain by binding to nicotinic and muscarinic acetylcholine receptors (nAChR and mAChR, respectively). The concentration of acetylcholine in the synaptic cleft is regulated by acetylcholinesterase, a membrane-bound enzyme that hydrolyzes acetylcholine, thereby terminating its synaptic activity [34,76]. According to existing studies, T. gondii infection is associated with increased acetylcholinesterase activity in the brain, leading to the enhanced hydrolysis of acetylcholine in the synaptic cleft and a corresponding decrease in its levels in the CNS. Given the proposed anti-inflammatory function of acetylcholine, this decrease may contribute to maintaining inflammatory processes and host defense mechanisms against the parasite. Such processes may underlie brain dysfunctions related to toxoplasmosis, including abnormalities in behavior, locomotion, memory, balance, and orientation [34,38]. Evidence further suggests that the cholinergic system is involved in ASD pathogenesis, as exhibited by reductions in neuronal nAChR in the brain cortex, defective acetylcholine signal transduction, lower baseline levels of extracellular acetylcholine, and abnormal acetylcholinesterase activity (Fig. 2) [76–78]. Thus, disruptions in the cholinergic system associated with toxoplasmosis may contribute to neuropsychiatric disorders that are also characteristic of ASD.
Gamma-aminobutyric Acid
Gamma-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the CNS, regulating the flow and timing of excitatory neurotransmission [33]. Beyond its role in neurotransmission, GABA is involved in various cellular processes, such as proliferation, differentiation, migration, synapse organization, axonal development, and neuronal death [79]. Glutamate decarboxylase (GAD), the key enzyme that catalyzes the conversion of glutamate to GABA at inhibitory synapses, exists in two isoforms—GAD65 and GAD67 [79,80]. Studies have shown that GAD65 and GAD67 levels are decreased in the brains of individuals with ASD, resulting in reduced GABA levels and enhanced glutamate concentrations [80,81]. Notably, T. gondii disrupts inhibitory GABAergic synaptic transmission [33,37], a process implicated in the pathogenesis of neuropsychiatric disorders such as ASD [80,81]. One parasite-induced alteration involves changes in the distribution of the GAD67 enzyme in the brains of individuals with toxoplasmosis. This alteration is associated with spontaneous seizures and may indicate GABAergic system deficits and a loss of neuronal inhibition (Fig. 2) [33,37]. Furthermore, T. gondii utilizes GABA as a carbon source for its metabolism. The intracellular localization of live parasites induces GABA secretion in parasitized dendritic cells (DCs), which facilitates parasite dissemination by stimulating DC motility. Thus, the parasite manipulates GABAergic signaling in DCs to enhance its dissemination and survival [33,79].
Glutamate
Glutamate, the primary excitatory neurotransmitter in the CNS, plays a crucial role in neuronal plasticity and cognitive function. However, excessive or unregulated glutamate levels can lead to neuroexcitotoxicity and neuronal cell death, processes implicated in the pathophysiology of several neuropsychiatric disorders, including ASD [39,80,82]. Glutamate is fully synthesized de novo within the CNS by astrocytes and neurons, which also contributes to the immune response against T. gondii. Astrocytes regulate glutamate levels in the CNS through uptake, release, conversion into glutamine, and synthesis from precursors, such as lactate, alanine, or α-ketoglutarate. Neurons convert glutamine to glutamate through the glutaminase enzyme [39]. Toxoplasmosis infection results in a substantial reduction in the astrocytic glutamate transporter, which is essential for clearing glutamate from the synaptic cleft following neurotransmission, leading to a significant increase in extracellular glutamate concentrations (Fig. 2) [37,39]. These findings indicate that abnormalities in glutamatergic neurotransmission caused by T. gondii may contribute to the neurophysiological mechanisms underlying ASD.
Gender
Sex differences have been observed in hosts with toxoplasmosis regarding susceptibility to infection, immune responses, alterations in brain neurotransmitter levels, and parasite-induced behavioral and personality changes [83–87]. Experimental studies have revealed that female mice are more susceptible to acute infection than male mice. Among those who survive and progress to chronic infection, females tend to have a higher cyst burden in the brain than their male counterparts [84,85]. Further immunological examination revealed differences in innate and adaptive immune responses—such as cytokine production, cell proliferation, and immune cell activity—which may explain the poor survival rates and higher cyst burdens in female mice [84,85]. Additional studies have highlighted the role of sex hormones as potential modulators of these sex-specific immune responses [88–90]. Notably, changes in neurotransmitter levels—believed to influence intermediate host behavior during T. gondii infection—also show sex-dependent patterns [83]. These sex-dependent effects on the essential molecular pathways of the host body suggest that T. gondii may play differing roles in the progression of ASD between males and females. Meanwhile, epidemiological studies consistently report a lower prevalence of ASD in females than males, possibly due to sex-specific genetic and hormonal influences, although the underlying mechanisms remain unclear [91–93]. Therefore, further research is needed to clarify these uncertainties and elucidate how T. gondii may differentially affect ASD progression across the sexes.
Genetics and Environmental Factors
ASD encompasses a group of distinct disorders that are strongly influenced by genetics and environmental factors, although the precise underlying causes remain unclear [94,95]. The heritability of ASD is approximately 61%–94% [96]. Twin and family studies report concordance rates for ASD diagnosis of approximately 98% in monozygotic twins and 53% in dizygotic twins, with lower concordance observed in nontwin siblings and markedly lower prevalence in the general population [94].
Environmental factors are also implicated in ASD risk, spanning prenatal, natal, and postnatal periods. These include maternal infection or depression during pregnancy, abnormal interpregnancy intervals (both short and long), increased maternal body mass index, prenatal exposure to radiation and sodium valproate, cesarean delivery, gestational age at birth, low birth weight, and measles, mumps, and rubella vaccination [94,95,97].
Conclusion
This review shows that T. gondii infection constitutes a significant risk factor for ASD. The changes induced by toxoplasmosis in the host—including the activation of humoral and cellular immune responses, the production of various cytokines, changes in neurotransmitter levels (e.g., serotonin, dopamine, acetylcholine, GABA, and glutamate), and the activation of enzymes such as IDO—may contribute to the development and progression of ASD.
Moreover, further intensive studies are necessary to clarify the effects of this parasite on the etiology and progression of ASD. Elucidating the mechanisms involved in host-parasite interactions and examining the role of parasite genotypes in these processes is essential to advancing our understanding of the association between toxoplasmosis and ASD. Given that various T. gondii strains exhibit varying pathogenicity and elicit distinct immune responses in the host, infections caused by these strains likely have differential impacts on the course of neurodegenerative and neurodevelopmental disorders, as suggested by recent studies [98,99]. It is hoped that the findings of this review will guide future studies aimed at reducing the burden of mental disorders globally.
Notes
Author contributions
Conceptualization: Khalilian A, Daryani A
Data curation: Mikaeili Galeh T, Nayeri T, Dodangeh S, Hosseininejad Z, Tanzifi A
Investigation: Mikaeili Galeh T, Nayeri T, Dodangeh S, Hosseininejad Z, Tanzifi A
Methodology: Khalilian A
Project administration: Mikaeili Galeh T, Daryani A
Supervision: Daryani A
Validation: Mikaeili Galeh T
Writing – original draft: Mikaeili Galeh T, Nayeri T, Dodangeh S, Hosseininejad Z, Tanzifi A
Writing – review & editing: Mikaeili Galeh T, Daryani A
Conflict of interest
The authors declare no conflict of interest related to this study.
Acknowledgments
The authors thank Khoy University of Medical Sciences for approving this research (approval No. 402000029) under the ethics code IR.KHOY.REC.1402.051.