Divergent long-terminal-repeat retrotransposon families in the genome of Paragonimus westermani
Article information
Abstract
To gain information on retrotransposons in the genome of Paragonimus westermani, PCR was carried out with degenerate primers, specific to protease and reverse transcriptase (rt) genes of long-terminal-repeat (LTR) retrotransposons. The PCR products were cloned and sequenced, after which 12 different retrotransposon-related sequences were isolated from the trematode genome. These showed various degrees of identity to the polyprotein of divergent retrotransposon families. A phylogenetic analysis demonstrated that these sequences could be classified into three different families of LTR retrotransposons, namely, Xena, Bel, and Gypsy families. Of these, two mRNA transcripts were detected by reverse transcriptase-PCR, showing that these two elements preserved their mobile activities. The genomic distributions of these two sequences were found to be highly repetitive. These results suggest that there are diverse retrotransposons including the ancient Xena family in the genome of P. westermani, which may have been involved in the evolution of the host genome.
INTRODUCTION
Transposable elements (TEs) are found to be inserted into the genomic DNAs of most organisms. According to their modes of transposition, they are classified into broad groups: retrotransposons (class I) and transposons (class II) (Finnegan, 1992). Transposons move directly as DNA in a 'copy and paste' or 'cut and paste' fashion, whereas retrotransposons expand via reverse transcription of an mRNA intermediate transcribed from the mobile element. At equilibrium, there are numerous copies of given TEs in a host genome ranging from just a few to tens, or hundreds, of thousands, and each host is likely to harbour many different types of TEs. Therefore, these elements may represent a major fraction of the genome, especially those of higher animals, including humans (Li et al., 2001), and plants (SanMiguel et al., 1996).
Retrotransposons are a common class found in eukaryotic genomes. They are categorized into two large groups, long-terminal-repeat (LTR) and non-LTR retrotransposons, on the basis of their overall structures. Recent phylogenetic analyses based on the amino acid sequences of Pol proteins have demonstrated that each of these two groups is composed of several distinct clades, members of which are thought to be tightly related one another in evolutionary terms (Malik et al., 1999; Malik and Eickbush, 1999, 2001). Evolutionary relationships among these retrotransposon clades, including retroviruses, are also suggested by these studies (Malik and Eickbush, 2001; Xiong and Eickbush, 1990).
LTR retrotransposons are classified into two main families, Pseudoviridae (corresponding to the traditional Ty1/copia group) and Metaviridae (Ty3/gypsy group). The Metaviridae are further split according to the presence of the env gene (genus Errantivirus) or its absence (genus Metavirus) (Pringle, 1999). However, the taxonomical classification of LTR retrotransposons are likely to be more complex due to the recent identifications of new subgroups such as Bel, Xena, and DIRS, which show different structural or sequential features from the existing elements (Malik and Eickbush 2001; Dalle Nogare et al., 2002).
The genomes of the platyhelminthes are generally small and are highly repetitive (Regev et al., 1998 and references therein), therefore, these organisms could be significant models for the study of the evolutionary roles of retrotransposons. However, few reports have been issued on the retrotransposons of platyhelminthes, except for those of Schistosoma spp. and Clonorchis sinensis (reviewed in Brindley et al., 2003). A lung fluke of carnivorous animals including human, Paragonimus westermani, is widely distributed in Asia (Blair et al., 1999). Chromosomal studies have shown that the natural populations of P. westermani in northeast Asia, such as Korea, China, Japan, and Taiwan, have three different levels of polyploidy in their genomic contents, i.e., di-, tri-, and tetraploidy (reviewed in Blair et al., 1999). It is well known that the expansion of retrotransposons influences chromosome evolution (O'Neill et al., 1998) and the expression profiles of host genes (Kidwell and Lisch, 1997), and that hybridization process between or within species can affect the mobile activity of retrotransposons (Zhao et al., 1998). In these regards, P. westermani is considered an useful model system in the elucidation of the degree of genomic alteration after hybridization and the resulting changes in gene expression patterns induced by retrotransposon expansion.
In the present study, we isolated and characterized retrotransposons in the genome of P. westermani by adapting a degenerate PCR method. Cloning and sequencing of the PCR products demonstrated that there are various retrotransposons belonging to divergent LTR retrotransposon families. The preserved mobile activities of certain elements are suggested by the presence of their mRNA transcripts. Genomic distribution patterns of these elements were also observed.
MATERIALS AND METHODS
Parasites and genomic DNA extraction
P. westermani metacercariae were collected from naturally infected crayfish in Haenam (2n) and Bogildo (3n), Korea (Park et al., 2001). Five months after infecting experimental dogs, adult worms were harvested from the lungs and were washed with physiological saline five times at 4℃. Worms were stored in liquid nitrogen until use. Frozen worms were ground in liquid nitrogen using a mortar and pestle, and DNA was extracted using a Wizard DNA Purification Kit (Promega, Madison, WI, USA), according to the manufacturer's instructions. The concentration of the extracted genomic DNA was measured by electrophoresis on an agarose gel using lambda DNA as a quantitative control.
Degenerate PCR and cloning
Candidate retrotranposon regions were amplified by degenerate PCR using genomic DNA of P. westermani obtained from Haenam, as a template. The primers D1 (forward direction, 5'-GTT/GTTIG/TTIG AT/GACIGGIG/TC-3') and D2 (reverse direction, 5'-ATIAGIAG/TA/GTCA/GTCIACA/GTA-3') matched to the sequences of protease (pr) and reverse transcriptase (rt), respectively, encoded in LTR retrotransposons were used in the reaction (Tristem, 1996). The reaction mixture included 50 ng of genomic DNA, 5 µM of primers, 0.2 mM each of dATP, dGTP, dCTP, and dTTP, and 1.25 U of Taq polymerase (Takara, Shiga, Japan) in a total reaction volume of 20 µl. PCR cycling parameters were as follows: 1 cycle of 4 min at 94℃; 35 cycles of 40 sec at 96℃, 40 sec at 48℃, and 1 min 30 sec at 72℃; followed by 1 cycle of 10 min at 72℃. The amplified PCR products were fractionated by electrophoresis on a 1.2% agarose gel and visualized by ethidium bromide staining. The products were totally cloned into pGEM-T Easy Vector (Promega) for nucleotide sequencing.
Sequence analysis
The nucleotide sequences were automatically determined using an ABI PRISM 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA) and a BigDye Terminator Cycle Sequencing Reaction Kit (Perkin Elmer Corporation, Foster City, CA, USA). To ensure the accuracy of the sequencing reactions, nucleotide sequences from both strands were determined. The nucleotide sequences obtained were used as queries in homology searches against the nonredundant database at the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/) by using BLAST (Altschul et al., 1997).
Southern blot hybridization
Five micrograms of genomic DNAs isolated from P. westermani were digested with restriction enzymes such as Acs I, Sac I, and Sfu I. After being fractionated through a 0.8% agarose gel, the restricted fragments were then blotted onto a nylon membrane (Hybond-N+; Amersham Pharmacia Biotech, Uppsala, Sweden) by capillary action in 10 × standard saline citrate (SSC). The blot was hybridized with retrotransposon-specific DNA probes enzymatically labeled by an ECL Direct Labeling Kit (Amersham Pharmacia Biotech). The labeling and hybridizing conditions followed the manufacturer's instructions. The hybridized membrane was washed twice in 6 M urea, 0.4% sodium dodecyl sulfate, and 0.1 × SSC at 42℃ for 20 min and then, twice in 2 × SSC at room temperature for 5 min. To detect signals, the membrane was exposed to Hyperfilm™ MP (Amersham Pharmacia Biotech) for 10 min after adding ECL detection reagents (Amersham Pharmacia Biotech).
RT-PCR
Total RNAs were extracted from whole bodies of P. westermani obtained from Haenam with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), as described by Chomczynski (1993). Specific primers were designed to match both ends of the retrotransposon-related sequences obtained from the P. westermani genome by the degenerate PCR. The RNAs and primers were used in RT-PCR to detect the mRNA transcripts of each retrotransposon. PCRs were conducted using a RNA PCR Kit (version 2.1; Takara), following the manufacturer's instructions. A cysteine protease gene identified in P. westermani (unpublished data) was used in the reaction as a positive control.
Phylogenetic analysis
RT sequences of almost all retrotransposons contain seven well conserved domains in terms of length and amino acids, which are intervened by hypervariable sequence segments (Xiong and Eickbush, 1990). The amino acid sequences from domains I to IV of RT, which can amplified by the degenerate primers, were aligned using ClustalX (Thompson et al., 1997). After optimizing the alignment with GeneDoc (Nicholas and Nicholas, 1997), a phylogenetic analysis was performed using the PHYLIP package (Felsenstein, 1993). The phylogenetic tree was displayed by TreeView (Page, 1996), and the statistical significances of branching points were evaluated by 100 random samplings of the input alignments using SEQBOOT. The RT sequences used in the phylogenetic analysis were CsRn1 (AY013569), Tom (CAA80824), ZAM (CAA04050), Gypsy (M12927), Ty3-2 (S53577), Athila (AB005248), Dea1 (Y12432), Sushi (AF030881), Ulysses (X56645), Osvaldo (AJ133521), Mag (S08405), Blastopia (Z27119), Cer1 (U15406), PAT (X60774), DIRS1 (M11340), Prt1 (Z54337), Pao (L09635), Ninja (AB042129), Tas (Z29712), Cer 11 (U41268), Ta1 (X13291), Copia (M11240), Ty1 (M18706), Penelope (U49102), Nep-TnB (AL329115), CRE1 (M33009), cauliflower mosaic virus (NC_001497), figwort mosaic virus (NC_003554), carnation etched ring virus (NC_003498), woodchuck hepatitis B virus (JDYLC2), hepatitis B virus (P12933), duck hepatitis B virus (AF493986), human immunodeficiency virus 1 (AAB50259), Rous sarcoma virus (NP_056886), and baboon endogenous virus (BAA89659). These sequences were obtained from the GenBank database of NCBI. Where possible, protein sequences were used directly from the database; otherwise, the sequences were predicted by translating ORF2.
RESULTS
Isolation of retrotransposon-related sequences
The genome of P. westermani was screened by PCR with the previously described degenerate primers (Tristem, 1996), to amplify the retrotransposon-related sequences. After cloning and sequencing, the nucleotide sequences of the 45 selected clones were used as queries in the homology searches against the nonredundant database of GenBank at NCBI using the tBLASTX algorithm. Of these, 26 sequences (58%) were found to have strong identities to those of Pol proteins encoded in the various retrotransposons at the amino acid sequence level. Comparisons of homology patterns and nucleotide sequences revealed that these sequences represented 12 different types of retrotransposons residing in the genome of P. westermani (Table 1). The homology value of 70% at the nucleotide level was adapted to verify the individuality of each sequence. In cases of retrotransposons retrieved with redundancy, considerable degrees of sequence divergence were observed among their clones, suggesting that these clones may have been amplified from different copies of a retrotransposon.

Sequence identities of retrotransposon fragments from the genome of P. westermani
The mobile activities of retrotransposons
For genomic expansion, retrotransposons should be transcribed into mRNA molecules by the host RNA polymerase and then, be reverse transcribed into double stranded cDNA by their own RT (Boeke et al., 1985). Therefore, the potential mobilities of retrotransposons can be primarily predicted by the presence of their mRNA transcripts. Thus, RT-PCRs were conducted using retrotransposon-specific primers and total RNAs extracted from the whole bodies of P. westermani adults. As shown in Fig. 1, the mRNA transcripts of two retrotransposons encompassing the sequences of clones Pw-d-23 and Pw-d-100, respectively, were detected, suggesting that these two elements are active in the present genome of P. westermani at the transcription level. The genomic distribution patterns of these elements were shown to be highly repetitive in the trematode genome. However, no difference in the distribution patterns of diploid and triploid worms was detected by the Southern blot analyses (Fig. 2).

Expression of retrotransposon sequences isolated from the genome of Paragonimus westermani. mRNA transcripts of the elements were amplified by RT-PCR with element-specific primers, using total RNAs extracted from whole bodies of Paragonimus westermani adults as a template. The cysteine protease gene was used as a control. Products were visualized by ethidium bromide staining after electrophoresis on agarose gels. M, 100-bp DNA ladder.
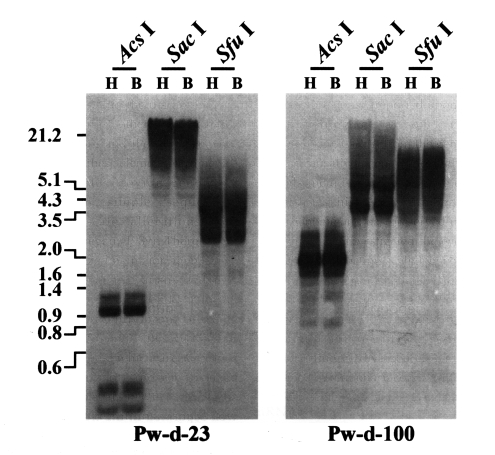
Distributions of CsRn1-like Pw-d-23 and Bel-like Pw-d-100 in the diploid (H) and triploid (B) genomes of Paragonimus westermani. Intragenomic distribution patterns of the retrotransposons were analyzed by Southern blot hybridizations with the genomic DNAs of each populations digested with several restriction enzymes. The positions of DNA size standards (in kb) are shown on the left.
Retrotransposon families in the genome of P. westermani
The amino acid sequences of RT from domains I to IV were aligned (Fig. 3) according to the conserved domain scheme of Xiong and Eickbush (1990). Since almost all retrotransposons isolated from the genome of P. westermani were degenerate forms, where possible, the amino acid sequences were determined from a consensus sequence of each type. Otherwise, the sequences were directly translated from each single clone after correcting the sequences to increase homology values with the RTs of other retrotransposons. The evolutionary positions of the trematode retrotransposons were estimated by a phylogenetic analysis of 47 retrotransposons, including all the sequences from this study and a number of other representatives of LTR-containing genetic elements (Malik and Eickbush, 2001; Dalle Nogare et al., 2002).

Multiple sequence alignment of retrotransposons isolated from Paragonimus westermani and their relatives. The amino acid sequences of domains from I to IV of reverse transcriptase (Xiong and Eickbush, 1990) are aligned. Sequence identities among these are highlighted by gray boxes. Parentheses in this figure show the numbers of amino acids between conserved domains.
A phylogenetic tree was constructed using maximum parsimony algorithm and rooted with a non-LTR retrotransposon, CRE1, based on the multiple sequence alignment. LTR-containing genetic elements are divided into eight distinct families, namely, Xena, Copia, hepadnavirus, Bel, DIRS, retrovirus, caulimovirus, and Gypsy, largely on the basis of amino acid conservations in their Pol sequences. The members of these eight families were well separated in the tree as shown in Fig. 4. The sequences of P. westermani included four members of Xena (Pw-d-5, -17, -28, and -45), two members of Bel (Pw-d-35 and -100), and six members of Gypsy (Pw-d-1, -2, -6, -8, -23, and -59). In addition, the six members of the Gypsy family seemed to be clustered into two distinct clades. It was apparent that Pw-d-1 and -23 were members of the CsRn1 clade previously detected in trematodes and insects (Bae et al., 2001; Copeland et al., 2003), whereas the positions of the other sequences appeared to be distinct from those of known clade members (Malik and Eickbush, 1999). The statistical significance of major branching nodes was well supported by a bootstrap analysis, and a similar clustering pattern was observed in the tree constructed using the UPGMA algorithm (data not shown).
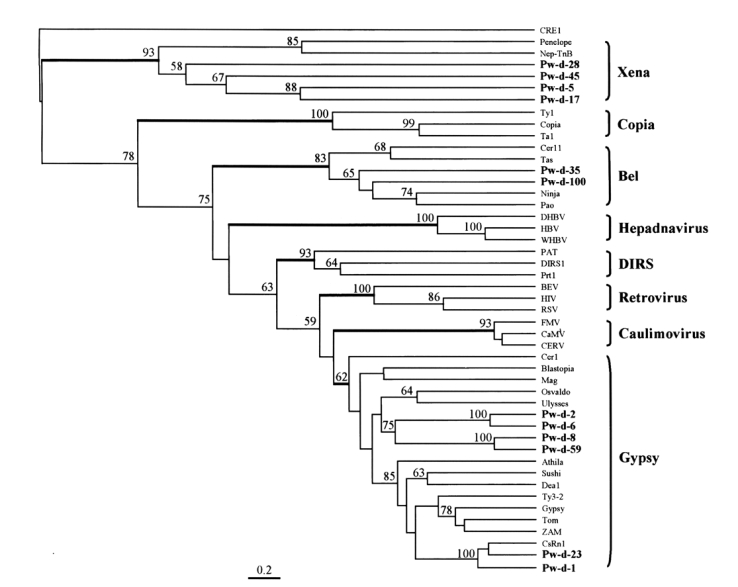
The relationship between retrotransposons of Paragonimus westermani and known retroelement families. This phylogenetic tree was based on the amino acids of domains from I to IV of reverse transcriptase and constructed employing the maximum parsimony algorithm implemented in PHYLIP. The statistical significance of each branching node is presented as a bootstrap value (100 replicates). The tree was rooted with a non-LTR retrotransposon CRE1. Names in bold are sequences used in this study.
DISCUSSION
Several properties of retrotransposons including their coding profiles (Tristem, 1996; Cook et al., 2000) and repetitivenesses (Blesa and Martínez-Sebastián, 1997; Drew and Brindley, 1997), and genomic polymorphisms introduced by retrotransposons (Abe et al., 1998; Bae et al., 2001) have previously been targeted for the selective isolation of retrotransposons. These approaches proved effective, and numerous novel elements were successfully identified. In the present study, we selected a degenerate PCR method based on their coding profiles, to comprehensively isolate retrotransposons residing in the genome of P. westermani. The screening procedure was very efficient and 58% of the cloned sequences encompassed retrotransposons. These sequences represented partial sequences of 12 different types, which were separately grouped into divergent families of LTR retrotransposons such as Xena (four types), Bel (two types), and Gypsy (six types).
Endogenous LTR-containing genetic elements (LTR retrotransposons) can be divided into large families of Xena, Bel, Copia, DIRS, and Gypsy according to amino acid conservation in their Pol proteins and differences in their overall structures (Malik and Eickbush, 2001; Xiong and Eickbush, 1990). The main difference between the Copia family and the others is the position of the integrase (IN) domain. It is found upstream of the RT/RNase H (RH) domains in the Copia family but downstream in the others. We could not isolate members of the Copia family in this study. The IN domain is the largest domain encoded in the Pol proteins of retrotransposons. Failure to retrieve the Copia sequences might result from difficulties in amplifying the larger fragments using our PCR conditions, because of the ubiquitous distribution shown by Copia members (Peterson-Burch and Voytas, 2002) and previous result of isolations performed with the same primers (Cook et al., 2000). Members of the DIRS family were not observed. It could not be concluded whether this was due to the absence of the DIRS family in the genome of P. westermani, as the distributions of DIRS across evolutionary taxa are largely unknown.
The Gypsy family is composed of eight well conserved clades (Malik and Eickbush, 1999), and an additional novel clade (CsRn1 clade) was recently detected (Bae et al., 2001). This clade included retrotransposons identified mainly in the genomes of trematodes and insects. Two elements (Pw-d-1 and -23) of P. westermani were found to belong to the CsRn1 clade by a phylogenetic analysis (Fig. 4), suggestive of the ubiquitous distributions of these members in the genomes of the lower animals. The phylogenetic analysis also showed that four clones assigned to the Gypsy family (Pw-d-2, -6, -8, and -59) formed one or two distinct clade(s) differently clustered from any known clade members of the family (Fig. 4).
Bel-like elements have been described in several insect species (Cook et al., 2000) as well as in nematodes (Felder et al., 1994; Bowen and McDonald, 1999). They have a typical genomic organization, similar to that of the Gypsy family. In spite of this structural similarity, the Bel family is likely to precede the Gypsy family as an important intermediate form during the evolutionary course of diverse reverse transcribing elements (Dalle Nogare et al., 2002). We detected two retrotransposons (Pw-d-35 and -100) related to members of the Bel family. Unlike those of the Gypsy family, these elements had relatively long interdomain sequences in RT (Fig. 3). The nucleotides corresponding to domain IV of RT were deleted in clone Pw-d-100. The retrotransposon encompassing the clone was shown to be active at the transcription level (Fig. 1). Thus, the sequence of clone Pw-d-100 might be amplified from a corrupted, inactive copy of the retrotransposon with a large deletion(s) in that region.
The evolutionary origin of LTR-containing genetic elements including retrovirus has been recently suggested by amino acid sequence analysis of RTs (Malik and Eickbush, 2001). Relating this model, Dalle Nogare et al. (2002) suggested that members of the Xena family are the most ancient retrotransposons, which diverged during an early evolutionary stage of LTR retrotransposons. Xena-like elements have been shown to be present in a wide range of taxa including insects, deuterostomes, and echinoderms, and have certain structural features in common (Dalle Nogare et al., 2002). However, information on Xena-like elements is restricted since only few studies have examined the full-units of retrotransposons belonging to the Xena family. In trematodes, one partial sequence of a Xena-related retrotransposon was detected in the EST database of Schistosoma mansoni (AI974952; Dalle Nogare et al., 2002). We isolated four partial sequences of Xena-like retrotransposons from the genome of P. westeramni (Fig. 4), suggesting that these elements are also ubiquitous in platyhelminthes.
The potential mobilities of retrotransposons isolated in this study were examined by testing for presence of their mRNA transcripts. At least two elements (Pw-d-23 and -100) were actively transcribed into mRNA in the nucleus of P. westermani (Fig. 1). With preserved mobile activities, these elements might have induced considerable degrees of genomic variations in the genome of P. westermani at the both intra- and inter-population levels. mRNA transcripts of the other clones were not detected in this analysis. Almost all clones retrieved contained heavily corrupted coding sequences, which means that these were degenerated, inactive variants. Even though we designed consensus primers for the RT-PCR, where possible, the highly substituted bases in our sequences make it difficult to exclude the potential mobilities of the other clones.
The hybridization process is regarded as one of the potent causal factors that increases the mobile activities of various retrotransposons (Evgen'ev et al., 1997; Labrador et al., 1999). The genomic distributions of two active Paragonimus retrotransposons (Pw-d-23 and -100) were not differentiated in diploid and triploid worms by Southern blot analyses with the pooled genomic DNAs of each population (Fig. 2). Retrotransposons randomly select target for their insertions in host genomes (Kido et al., 1995), which results in heterogeneous integration patterns among individuals. Genomic loci having retrotransposons individually inserted, if any, would not be easily detected by Southern blotting with the pooled genomic DNAs. More detailed examinations in individual genomes of P. westermani, such as inter-retrotransposon amplified polymorphism analysis (Manninen et al., 2000), would be needed to determine the degree of genomic alterations caused by the expansion of active retrotransposons.
Only limited data on LTR retrotransposons are available for the phylum Platyhelminthes (Brindley et al., 2003). Model organisms commonly used in studies upon the biological implications of retrotransposons, such as Drosophila melanogaster, Arabidopsis thaliana, and Saccharomyces cerevisiae, have simple genomes that harbor repetitive elements at low copy numbers. This makes it difficult to estimate the actual significance of retrotransposons in the complex genomes of higher animals. The present study of a trematode provides advantages for the study of retrotransposons in its small but complex genome. Moreover, the results obtained would be helpful to broaden our understanding on the dynamic evolution of retrotransposons.
Notes
This study was supported by a Samsung grant, #SBRI BA2-006.