Possible Role of Heme Oxygenase-1 and Prostaglandins in the Pathogenesis of Cerebral Malaria: Heme Oxygenase-1 Induction by Prostaglandin D2 and Metabolite by a Human Astrocyte Cell Line
Article information
Abstract
Astrocytes are the most abundant cells in the central nervous system that play roles in maintaining the blood-brain-barrier and in neural injury, including cerebral malaria, a severe complication of Plasmodium falciparum infection. Prostaglandin (PG) D2 is abundantly produced in the brain and regulates the sleep response. Moreover, PGD2 is a potential factor derived from P. falciparum within erythrocytes. Heme oxygenase-1 (HO-1) is catalyzing enzyme in heme breakdown process to release iron, carbon monoxide, and biliverdin/bilirubin, and may influence iron supply to the P. falciparum parasites. Here, we showed that treatment of a human astrocyte cell line, CCF-STTG1, with PGD2 significantly increased the expression levels of HO-1 mRNA by RT-PCR. Western blot analysis showed that PGD2 treatment increased the level of HO-1 protein, in a dose- and time-dependent manner. Thus, PGD2 may be involved in the pathogenesis of cerebral malaria by inducing HO-1 expression in malaria patients.
INTRODUCTION
Astrocytes are the most numerous cell types in the central nervous system (CNS). They provide structural, trophic, and metabolic support to neurons and modulate synaptic activity [1]. Astrocytes were used in various studies involving in neurological diseases [2-5]. These cells have the most direct interaction with vasculature, and therefore their endfeet are in contact with cerebral endothelial cells (CECs) [6,7]. In vitro and in vivo studies support the role of astrocytes in controlling blood-brain-barrier (BBB) maintenance and regulation through their interaction with CECs [8-12]. The neurological dysfunction caused by breakdown of BBB thereby allowing compounds, such as histamine and reactive oxygen species (ROS) to enter the brain, may lead to the pathological changes in the brain [13].
Malaria is a worldwide protozoan infection [14], and most malignant malaria is caused by Plasmodium falciparum. Cerebral malaria is one of the most severe complications of P. falciparum infection [15,16]. The parasite does not enter the brain parenchyma, but staying in the intravascular circulation, which is responsible for changes in the BBB. Cerebral malaria is characterized by a sequestration of parasitized red blood cells in the brain microvasculature. The sequestration of these parasitized erythrocytes could result in activation of cerebral endothelial cells [17]. Perivascular macrophages (perivascular cells) are amongst the first cells to encounter proteins leaking across a disrupted BBB, and as a consequence of activation or phagocytosis, they may secrete a wide range of pro-inflammatory and neuroactive mediators which could influence local neuronal functions. It has been reported that, during cerebral malaria, the cerebral parenchyma is not largely affected, and local events occur within and around the cerebral microvasculature [18]. The molecular basis underlying cerebral malaria, nevertheless, remains unclear.
Recently, heme oxygenase (HO) has been proposed as one of the factors that play significant roles in the pathogenesis of falciparum malaria complication [19,20]. It is a microsomal enzyme which exists in 2 isoforms, i.e., HO-1 and HO-2. Heme oxygenases are rate-limiting enzymes in heme catabolism to generate biliverdin IXα/bilirubin IXα, carbon monoxide, and ferrous iron [20-22]. The expression levels of HO-1 is inducible or repressible, depending on cell types or cellular microenvironments, but expression levels of HO-2 are fairly constant [20,22]. The main regulation of heme catabolism is, therefore, determined by the balance between induction and repression of HO-1.
HO-1 levels in the brain were studied in Alzheimer's (AD), Parkinson's (PD), and infectious diseases, including malaria [23,24]. It has been proposed that the mechanisms for the fine-tuning of HO-1 expression may involve the polymorphic (GT)n sequence (n = 15-40) located in the human HO-1 gene promoter [23,25]. We have recently reported that the short (GT)n repeats (n < 28) in the HO-1 gene promoter are associated with higher incidence of cerebral malaria in the Karen ethnic minority group who live near the border between Myanmar and Thailand [26].
Prostaglandins (PGs) originate from the degradation of membrane arachidonic acid by cyclooxygenases (COX-1 and COX-2). The prostaglandin actions in the nervous system have been suggested to play a significant role in neurodegenerative disorders [27] and to be involved in various symptoms associated with parasitic diseases [28]. PGD2 is a major prostanoid produced in the brain and is involved in the regulation of sleep and pain responses [29,30]. Lipocalin-type PGD synthase (L-PGDS) catalyzes the isomerization of PGH2 to produce PGD2, and is mainly responsible for production of PGD2 in the brain [30,31]. PGD2 exerts its actions through the G protein coupled receptors DP1 and chemoattractant-homologous receptor expressed on Th2 cells (CRTH2, also known as DP2) which are expressed in various cell types [32-34].
In 1998, Kubata et al. [28] have shown that the cell homogenates of P. falciparum contain the activity that produces PGD2 and PGE2 after incubation with arachidonic acid. Notably, PGD2 was shown to be the predominant accumulate in the culture medium of the parasitized erythrocytes after treatment with arachidonic acid [28]. Moreover, PGD2 was maintained at a higher level in the serum of falciparum malaria patients than the control serum [35]. We previously reported that PGD2 and 15d-PGJ2, a minor species of PGD2 metabolites, were found to increase HO-1 gene promoter, mRNA levels, and protein levels in retinal pigment epithelial cells that may associate with the pathogenesis of malarial retinopathy in malaria patients with cerebral complication [36,37].
In the present study, we provide additional data to support the link between HO-1 and pathogenesis of cerebral malaria through the induction by PGD2 in human astrocytes.
MATERIALS AND METHODS
Cell culture
The human astrocyte cell line, CCF-STTG1 was purchased from American Type Culture Collection (CRL-1718, ATCC, Virginia, USA). CCF-STTG1 cells were cultured in RPMI 1640 medium, supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and antibiotics (100 U/ml penicillin and 0.1 mg/ml streptomycin). To examine the effects of PGD2 and 15d-PGJ2 (Cayman Chemicals, Michigan, USA) on the expression levels of HO-1 protein and mRNA, CCF-STTG1 cells were grown to 70-80% confluence before they were incubated with vehicle (ethanol), PGD2 or 15d-PGJ2.
Western blot analysis
Cells were lysed in the buffer containing 20 mM Hepes (pH 7.9), 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1.5 mM MgCl2, 1 mM DTT, 0.1% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, Missouri, USA) and 0.5% Nonidet P-40 (Fluka, Buchs, Switzerland) [36]. The resultant lysates were centrifuged at 15,000 g for 5 min at 4℃ after incubation for 15 min, and then supernatants were collected. Lysate supernatant proteins (30 µg per lane) were fractionated by SDS-PAGE and blotted to a nitrocellulose membrane (Hybond ECL™, Amersham, New Jersey, USA) in buffer containing 20% methanol, 48 mM Tris, 39 mM glycine, and 0.037% SDS. The membranes (Western blots) were treated 1-2 hr in Tris-buffered saline (TBS), containing 5% non-fat dried milk, and were washed 3 times each for 10 min in TBS with 0.1% Tween 20 (TBS-T) at room temperature. The proteins were probed with HO-1 (SPA-895, StressGen Biotechnologies) and HO-2 (SPA-897, StressGen Biotechnologies, Michigan, USA) antibodies, and β-actin antibody (Sigma) at a dilution of 1 : 1,000 for 1 hr at room temperature. For detection of HO-1 and HO-2 proteins, the blots were incubated with alkaline phosphatase (AP)-goat anti-rabbit antibodies (ZyMax™, Invitrogen, California, USA), and β-actin protein was incubated with AP-goat anti-mouse antibodies (ZyMax™, Invitrogen). The color was developed by incubating the blot in the developing solution, BCIP/NBT (AMRESCO, Ohio, USA).
RNA extraction and RT-PCR
Total RNA was extracted from treated CCF-STTG1 cells using RNeasy® Mini Kit (Qiagen, Hilden, Germany). Total RNA was transcribed to cDNA using Omniscript® RT Kit (Qiagen). Then, HO-1 cDNA levels were determined by RT-PCR. Primers were designed according to the published cDNA sequences for human HO-1 [38] and HO-2 [39]. The forward 5'-CAGGCAGAGAATGCTGAG-3'and the reverse 5'-GCTTCACATAGCGCTGCA-3' primers for HO-1 were used to amplify a 271-bp fragment, and the forward 5'-ATGTCAGCGGAAGTGGAA-3'and the reverse 5'-GGGAGTTTCAGTGCTCGC-3'primers for HO-2 were used to amplify a 533-bp fragment. The house-keeping gene, glyceraldehyde-3-phosphate dehydrogenase, was also amplified using the following primers 5'-TGAAGGTCGGAGTCAACGGATTTG-3'and 5'-GCGCCAGTAGAGGCAGGGATGATG-3', yielding a 628-bp product [40].
The cDNA template were amplified with an initial hold for 5 min at 95℃, followed by 40 cycles of denaturation at 95℃ for 15 sec, and annealing/extension at 60℃ for 60 sec in iCycler iQ real time PCR (Bio-Rad, California, USA). Relative HO-1 mRNA and HO-2 mRNA expression were obtained by dividing the intensity value for each sample with 0-hr untreated control culture cells, which reflected basal expression level (normalized to 1.0) by using glyceraldehyde-3-phosphate dehydrogenase for standardization. Data were analyzed in duplicate.
RESULTS
Effects of PGD2 on the expression of HO-1 and HO-2 proteins in CCF-STTG1 cells
We performed the time-course studies of the effects of PGD2 on the expression of HO-1 and HO-2 proteins in CCF-STTG1 at 3, 6, 12, and 24 hr, and dose-response studies of PGD2 at a final concentration of 0.5, 1, 5, or 10 µM in CCF-STTG1 cells. Western blot analysis revealed that the expression of HO-1 protein was induced in a dose-dependent manner by PGD2 at a final concentration of 5 and 10 µM after a 24-hr treatment (Fig. 1A). The apparent induction of HO-1 protein was detected at 12 and 24 hr with the concentration of PGD2 5 and 10 µM. In contrast, the expression level of HO-2 protein was unchanged or did not increase after a 24-hr treatment with the highest concentration of PGD2 (10 µM). The expression of β-actin, an internal control was unchanged. These results indicate that PGD2 induces the expression of HO-1 protein in CCF-STTG1 cells.
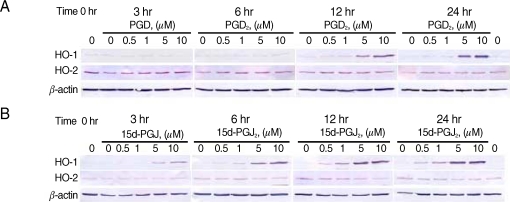
Effects of PGD2 and 15dPGJ2 on HO-1 and HO-2 proteins in CCF-STTG1 cells. CCF-STTG1 human astrocyte cells were treated with PGD2 (A) or 15d-PGJ2 (B) at final concentration 0.5, 1, 5, or 10 µM for the indicated time from 3 to 24 hr and then harvested for preparation of proteins. Shown are the Western blots used for HO-1 and HO-2 protein. Each lane contained 30 µg proteins prepared from CCF-STTG1 cells. A bottom panel shows β-actin as an internal control. The data shown are from 1 of 2 independent experiments.
Effects of 15d-PGJ2 on the expression of HO-1 and HO-2 proteins in CCF-STTG1 cells
It has been well established that 15d-PGJ2 increases transcription of the HO-1 gene [36,41,42]. We, therefore, performed a similar experiment with 15d-PGJ2 for comparison. As expected, 15d-PGJ2 induced the expression of HO-1 protein in a dose-dependent and time-dependent manner in CCF-STTG1 cells (Fig. 1B). The apparent induction of HO-1 was detected after a 6-hr treatment with 15d-PGJ2 (5 and 10 µM) (Fig. 1B). The onset of the HO-1 induction with 15d-PGJ2 was earlier than that with PGD2. HO-2 as well as β-actin protein levels remained unchanged after treatment with 15d-PGJ2.
Increased expression of HO-1 mRNA in CCF-STTG1 cells treated with PGD2 and 15d-PGJ2
We next performed the time-course and dose-response effects of PGD2 and 15d-PGJ2 on the expression of HO-1 mRNA in C CF-STTG1 cells using RT-PCR. The expression of HO-1 mRNA was induced by PGD2 or 15d-PGJ2 in a dose-dependent manner (Fig. 2). The expression levels of HO-1 mRNA were increased after 6 hr of the treatment with PGD2 of 5 µM or 10 µM, and continuously increased at maximum levels after 12 hr (Fig. 2A). For 15d-PGJ2, the expression levels of HO-1 mRNA were detected at 3 hr of treatment (Fig. 2C). The maximum induction of HO-1 mRNA was achieved after 6 hr of treatment, but it was decreased at 24 hr (Fig. 2C). In contrast, HO-2 mRNA levels remained unchanged in both of induction (Fig. 2B, D). The induction profiles of HO-1 mRNA were in good agreement with those of HO-1 protein (Fig. 1).

Expression of HO-1 mRNA by PGD2 and 15dPGJ2 in CCF-STTG1 astrocyte cells. CCF-STTG1 cells were treated with PGD2 or 15d-PGJ2 at final concentration 0.5, 1, 5, or 10 µM for the indicated time from 3 to 24 hr and then harvested for RNA preparation. cDNA were prepared for RT-PCR using Platinum® SYBR® Green qPCR SuperMix-UDG cocktail (Invitrogen). Shown are representative of the relative HO-1 mRNA and HO-2 mRNA expression of PGD2 (A, B) or 15d-PGJ2 (C, D) induction. The data were obtained by dividing the intensity value for each sample with 0-hr untreated control cells, which reflected a basal expression level.
DISCUSSION
The astrocytes are known as cells surrounding cerebral endothelial cells [43] that influence the BBB function [12,44,45] by releasing cytokines and other soluble factors which may induce adverse responses in surrounding neuronal tissues [46,47]. In addition, astrocytes have the property to synthesize prostaglandins [48,49].
The endothelial cells also have a reciprocal inductive influence on astrocytes [50,51]. Therefore, the astrocyte-endothelial interaction may be involved in the pathogenesis of encephalopathy caused by P. falciparum. Most of the antimalarial agents used in the treatment of cerebral malaria (quinine, artemether, artesunate, and mefloquine) have been reported to cause neurotoxicity [52-58]. The integrity of the BBB, including the functionality of the cerebral efflux proteins, e.g., p-glycoproten (P-gp), multidrug resistant protein-1 (MRP1), or breast cancer resistant protein (BCRP) are therefore important factors in cerebral malaria to control the cerebral transport and the neurotoxicity of these drugs [59-62]. Nevertheless, little information is available regarding the effects of alteration of BBB integrity in cerebral malaria as well as interactions of antimalarial agents on antimalarial toxicity [14,17,63,64].
The present results showing that, the treatment of astrocyzte cells CCF-STTG1 with exogenous PGD2 consistently induced HO-1 expression, is in agreement with our recent report in retinal pigment epithelial cells [36]. Endogenously produced PGD2 by astrocyte cells may contribute to the appropriate expression of HO-1 in the brain. The induction profile of HO-1 expression with PGD2 is essentially similar to that with 15d-PGJ2, although the onset of the induction by PGD2 is later than by 15d-PGJ2. It remains to be explored whether PGD2 itself or a specific PGD2 metabolite, other than 15d-PGJ2, is responsible for the induction of HO-1 expression. Apart from astrocyte cells themselves, falciparum parasites may also release PGD2 [28,48] that may hasten the expression of HO-1. This consequence additively results in the increase in HO-1 activity to catalyze heme to end products, especially, iron. Malaria growth and proliferation depend on iron supply from host cells, such as endothelial cells or neuron cells near the sequestration site. Therefore, PGD2 might enhance the growth of parasites by modulating iron availability from the host. The stimulation of HO-1 by PGD2 and the metabolite 15d-PGJ2 observed in this study is critical since excessive heme degradation may result in toxic levels of iron similar to that by carbon monoxide, and bilirubin/biliverdin.
Detailed studies are underway in order to provide evidence to definitely conclude on the prostaglandin-mediated stimulatory effects on HO-1 and its role in malaria pathogenesis. Preliminary studies showed significant findings. These include investigation of (i) the mechanism of PGD2-mediated induction of HO-1 expression in pigment epithelial cell lines (ARPE-19 and D407 RPE) in vitro, (ii) the production of PGD2 in P. falciparum culture in vitro, (iii) the growth inhibitory effects of the antagonists of PGD2 receptors (DP1 and DP2 antagonists) on HUVEC cells when cocultured with P. falciparum in vitro, and (iv) the association between PGD2 levels and malaria pathogenesis in patients. The knowledge on the link of the polymorphism of GT repeat of HO-1 gene promoter in modulating the enzyme activity of HO-1 and susceptibility to severe malaria may be exploited for further development of new drugs acting as inhibitors of HO-1 to prevent the progression to severe cerebral malaria.
ACKNOWLEDGEMENTS
This work was supported by Thammasat University and The Commission on Higher Education, Ministry of Education of Thailand.