Changes in enzyme activity and expression of DHFR of Toxoplasma gondii by antifolates
Article information
Abstract
The responses to antifolates of Toxoplasma gondii were investigated by measuring the dihydrofolate reductase (DHFR) activity, quantity of DHFR mRNA, and single-strand conformational polymorphism (SSCP) pattern. Pyrimethamine (PYM) and methotrexate (MTX) were tested as antifolates. When T. gondii was treated with PYM, the viability was decreased by the increasing concentration of PYM, DHFR activity tended to increase as the passage proceeded, and the quantity of mRNA expressed was also increased according to passages. The viability of T. gondii was decreased by the increasing concentration of MTX, but it was maintained over 40% up to 100 µM MTX. DHFR activity was 77.4% in the 1st passage (1 µM). 82.2% in the 4th passage (10 µM), and 141.3% in the 7th passage (100 µM). But no changes were detected in SSCP pattern of T. gondii exposed to PYM and MTX, both. These results suggested that the response of T. gondii to PYM was regulated by transcriptional level and that, in MTX, the viability of T. gondii was derived from increasing DHFR activity.
INTRODUCTION
The protozoan parasite Toxoplasma gondii is a ubiquitous pathogen long known for Its congenital infections (Remington and Desmonts, 1989). More recently, T. gondii has achieved considerable notoriety as an opportunistic infection afflicting AIDS patients (Luft and Remington. 1992).
Traditional treatment of toxoplasmosis involves blocking the folate metabolic pathway (Thaithong el al. 1992). Dihydrofolate reductase (DHFR) and thymidylate synthase (TS) take part in the folate metabolic pathway that supplies dTMP for DNA synthesis. In protozoa. DHFR and TS are on the same polypeptide chain: the DHFR domain is on the amino terminus, the TS domain is on the carboxyl terminus (Santi and Danenberg, 1984). The bifunctional DHFR-TS appears to be optimized for continuous dTMP synthesis. Co-transcription and co-translation of the two enzymes may provide an important element of coordinate control (Ivanetich and Santi. 1990). For that reason, DHFR and TS are targets for chemotherapy, as inhibition of either enzyme disrupts the dTMP cycle and results in the thymineless death of parasites. DHFR has been successfully used as a drug target in the treatment of diseases caused by protozoa, such as malaria. Pneumocystis carinii pneumonia, and toxoplasmosis (Garette et al., 1989). But antifolate resistant strains of other protozoa are found frequently, most of which have one or two point mutations in the DHFR domain (Reeder et al., 1996).
We investigated the concrete overcoming mechanism of T. gondii to permissive concen-trations of antifolates. For studying the responses in the translational level, DHFR activity was measured in normal RH strain and T. gondii treated with pyrimethamine and methotrexate. For transcriptional level, the density of DHFR and P30 gene was scanned after RT-PCR amplification. Single-strand conformational polymorphism (SSCP) was also performed to detect the changes of genomic DNA.
MATERIALS AND METHODS
Maintenance of host cells and T. gondii
U937 cells (human neoplastic leukemia cell line, ATCC CRL 1593) were cultured in Dulbecco's Modified Eagle Medium (DMEM, Gibco BRL Co.) supplemented with 10% fetal bovine serum (FBS. Gemini Co.). Toxoplasma gondii (RH srtain) were maintained by peritoneal passage in ICR mice.
Treatment of antifolates
Pyrimethamine (PYM, 0.001-10 µM) and methotrexate (MTX. 0.05-100 µM) were tested as antifolates. The growth rate was monitored by the uptake of [3H]-uracil (Dupont, 1 mCi/ml) uptake (Pfefferkorn and Pfefferkom, 1977) with the control without antifolates. And every 3rd passage, the concentration of antifolates was increased by 10 fold up to permissive concentration.
DHFR activity
Protein concentration of the crude extract of PYM and MTX treated RH was measured by the Bradford method using BSA as a standard (Bradford, 1976). DHFR activity was determined spectrophotometrically by monitoring the decrease in the absorbance at 340 nm, 25℃ in a reaction mixture containing Tris-HCl (50 mM, pH 7.6), β-mercaptoethanol (100 mM), KC1 (150 mM), NADPH (100 µM), DHF (100 µM) and EDTA (1 mM). The reaction was initiated by addition of DHF (Hillcoat et al., 1969). The decrease in A340 was monitored by spectrophtometer (8450A UV/VIS, Hewlett Packard Co.). The specific activity of the enzyme was calculated as mg-1min-1. The specific activity of normal RH was calculated at 100%.
Quantitation of DHFR mRNA by RT-PCR
DHFR mRNA was separated by Oligotex Direct mRNA micro kit (Qiagen 72012) to synthesize cDNA by reverse transcriptase (1st strand cDNA synthesis kit, Boehringer Mannheim). DHFR gene was amplified using the primers designed according to the sequence of Roos (1993), using upstream primer: 5'-CCATCGATATGCAGAAACCGGTGTGTCT-3' and downstream primer: 5'-CCATCGATACAAAGTCGTAGGGTACCCC-3'. To calculate the expression ratio, the major surface protein (P30) fragments of T. gondii was amplified with the same substrate using upstream primer: 5'-GCTCTAGATCACGCGACACAAGCTGC-3' and downstream primer: 5'-GCTCTAGATGTCGGTTTCGCTGCAC-3' (Nam et al., 1996) with restricted cycles in a programmable thermal controller (PTC-100, MJ Research Inc.). PCR product was electro-phoresed on a 1.5% agarose gel at 70 rnA for 40 min. The density of DHFR gene and P30 gene was measured by a video system (Eagle Eye II, Stratagene Co.), then the ratio of DHFR/P30 was calculated (Wang et al., 1989).
Detection of mutation in DHFR gen
Genomic DNA was obtained to use as substrate for PCR (Sambrook et al., 1989). The reactions were amplified as follows: initial denaturation for 30 sec at 95℃ and 29 cycles with denaturation at 95℃ for 1 min, annealing at 56℃ for 1 min, and extension at 72℃ for 2 min. The entire 1.73 kb of T. gondii DHFR gene was amplified using the same primers described above. After confirmed in 1.5% agarose gels, each PCR product was used as substate for the 2nd PCR by the primer sets in Table 1 for optimum separation in SSCP. During the reaction, 35S-dATP was incorporated, and PCR products were separated on 6% acrylamide gel under 500-800 V (30 mA) for 3-4 hrs. The gel was exposed to X-ray film to detect mutation (Dean and Gerrard, 1991). Plasmodial DHFR mutant gene M2M3 (Ser36 → Arg, Thr83 → Asn) was used as a positive control (Robert etal, 1993).

Primer sets to amplify the exon parts of DHFR genomic DNA of T. gondii
RESULTS
Both antifolates inhibited the growth of T. gondii with a little different pattern. When T. gondii was treated with PYM from 0.001 µM to 10 µM, the [3H]-uracil uptake ratio to control was decreased as the PYM concentration was increased (Fig. 1A). At the concentration less than 0.05 µM, the ratio was decreased rapidly by 74.4% from 97.7% at 0.001 µM to 23.3% at 0.05 µM. At more than 5 µM PYM, the ratio was decreased slowly, but maintained in 10%. When treated with MTX, the ratio was decreased slowly, but maintained over 40% from 5 µM to 100 µM MTX (Fig. 1B). It was estimated that the permissive concentration of PYM was up to 0.1 µM for the passage of T. gondii. And in case of MTX, concentration up to 100 µM was permissive for the passage.

Growth of T. gondii treated with various concentrations of PYM (A) and MTX (B). [3H]-uracil uptake was counted and calculated as % to normal RH.
DHFR activity with uracil uptake to control by the passage was described in Fig. 2. That of normal RH was 0.23 mg-1min-1 (100%). At 0.01 µM PYM, DHFR activity ratio to control was 33.9% in the 1st passage. 47.0% in the 2nd passage, and 47.4% in the 3rd passage. After increasing the PYM concentration to 0.1 µM, DHFR activity was recovered to 63.5% in the 4th passage (Fig. 2A). During the passage, the profile of uracil uptake ratio was followed to the DHFR activity except the 2nd passage that increased to 135.2%. In the passage under MTX treatment, the enzyme activity was increased slowly from 77.4% in the 1st passage with 1 µM MTX, 82.2% in the 4th passage with 10 µM, and to 141.3% in the 7th passage with 100 µM (Fig. 2B). Uracil uptake showed a similar pattern with DHFR activity.
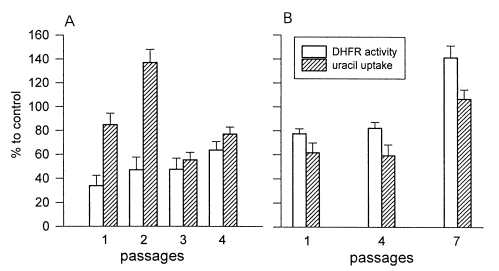
DHFR activity and uracil uptake of T. gondii with PYM (A) and MTX (B). A: passage 1, 2, and 3 were treated with 0.01 µM PYM and passage 4 with 0.1 µM. B: passage 1, 1 µM MTX; passage 4, 10 µM; and passage 7, 100 µM.
The ratio of DHFR/P30 mRNA expressed was obtained indirectly by RT-PCR of each mRNA. In normal RH, the ratio of DHFR/P30 mRNA expression was 0.64 under condition used. There were quantitative changes of DHFR mRNA from the 1st passage to the 4th passage with PYM. The ratio of DHFR/P30 was decreased by 0.5, 0.51, 0.49, and 0.53 in 0.001 µM, 0.005 µM, 0.01 µM, and 0.05 µM PYM in the 1st passage, respectively. That of DHFR/P30 was increased by 0.78, 1.29, 1.95, and 2.92 in the 4th passage of which the concentration of PYM was increased 10 fold to 0.01 µM (PI), 0.05 µM (P2), 0.1 µM (P3), and 0.5 µM (P4) (Fig. 3A). The ratio of DHFR/P30 was 1.14, 1.45, 2.20, and 0.75 in 0.05 µM, 0.5 µM, 5 µM, and 50 µM MTX in the 1st passage, but changed irregularly as 0,7, 2.57, 1.10 and 0.31 after increasing the concentration to 0.5 µM (Ml), 5 µM (M2), 50 µM (M3), and 100 µM (M4), respectively (Fig. 3B).
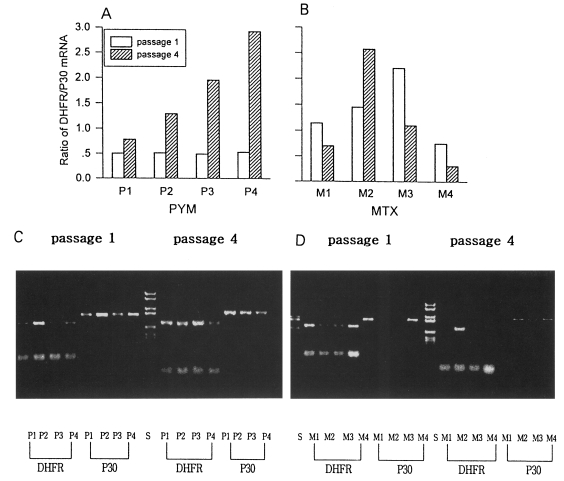
Ratio of DHFR/P30 mRNA expression of T. gondii by RT-PCR. A. Shifting of the concentration of PYM: PI, 0.001 → 0.01 µM; P2, 0.005 → 0.05 µM; P3. 0.01 → 0.1 µM; and P4, 0.05 → 0.5 µM. B. MTX: Ml, 0.05 → 0.5 µM; M2, 0.5 → 5 µM; M3, 5 → 50 µM; and M4, 50 → 100 µM. C (PYM) and D (MTX): DHFR and P30 DNA amplified by RT-PCR in 1.5 % agarose gel.
In SSCP pattern of DHFR exon fragments of T. gondii exposed to PYM and MTX showed no differences comparing to that of normal RH. This indicated that there were no changes of DHFR gene in the genomic level when exposed to both PYM and MTX (Fig. 4A & B).

SSCP pattern of DHFR exon fragments of T, gondii treated with PYM (A) and MTX (B). B1/B2 and B3/B4 : primer sets in Table 1 (DNA fragments from other primer sets not shown). A: 1, normal RH; 2, M2M3 (positive control); 3. PI (0.001 µM PYM); 4, P2 (0.005 µM); 5, P3 (0.1 µM); and 6, P4 (0.05 µM). B: 1, M2M3; 2, normal RH; 3, passage 1; 4, passage 2; 5, passage 3 (1 µM MTX); 6, passage 4 (10 µM); and 7, passage 7 (100 µM).
DISCUSSION
Both PYM and MTX induced different overcoming responses in T. gondii such as changes in DHFR activity and expression of DHFR mRNA. By PYM, T. gondii regulated the expression of DHFR mRNA to overcome the drug. And the over-expression of mRNA was dose dependent as shown in Fig. 3. After shifting to 10-fold from various concentrations of PYM as in PI, P2, P3 and P4, the quantity of mRNA was increased to 1.56, 2.47, 4.00 and 5.54 folds, respectively. But in case of MTX treatment, T. gondii regulated the DHFR activity rather than expression of DHFR mRNA, although the pattern of the latter was not defined clearly. DHFR activity was decreased by 77.4% in the 1st passage (1 µM), after then increased by 141.7% in the 7th passage (100 µM), Totally the responses were in the boundary of protein level and transcriptional level, not in genomic level.
Although MTX is one of strong DHFR inhibitors, it was difficult to induce the changes in genomic level with these concentrations except by increasing DHFR activity in this study. In T. gondii, although it was not the same pattern, the reaction to PYM and MTX was similar to that of Leishmanial major. MTX was much less effective in culture against T. gondii than its potency in vitro suggested it might be (Mack and Mcleod, 1984).
Resistance to DHFR inhibitors in various parasites has been shown to arise by diverse mechanisms, including gene amplification, point mutations at the hot spot of the gene, and alteration of drug uptake. When treated with a lethal dose of an antifolate that is an analogue of DHF or DHFR inhibitor, the parasites are dealt a fatal blow. If the concentration of antifolates is less than lethal dose, they change their survival strategies according to those conditions. They inhibit uptake of drugs, and quickly expell drugs from cells, or increase the quantity of enzyme, and induce conformational changes of structure of enzyme to inhibit the binding of antifolates (Wellem, 1991). In L. major, resistant-antifolate was derived from DHFR gene amplification. Antifolate resistant strains of L. major amplify the DHFR-TS gene in extrachromosomal circular DNAs which is correlated with enzyme overproduction, R region (Beverly, 1991). It appears that drug resistance in parasitic protozoa may be mediated by gene amplification (Coderre et al., 1983). Foote (1990) reported that amino acids involved in cyclo-guanil resistance differed from those involved in PYM resistance. Others have recently developed transformation systems capable of both transient and stable expres-sion of recombinant transgenes in T. gondii (Soldati and Boothroyd, 1993). Selection with PYM permits the isolation of stable clones harboring the mutant DHFR-TS gene at high frequency. Depending on the particular drug resistance allele chosen and conditions of selection, it is possible to select for the amplified transgenes or single copy chromo-somal insertions (Peterson etal, 1988).
In PYM-resistant strains of Plasmodium falciparum, all have a common point mutation in the DHFR coding sequence which causes decreased binding of the folate analogue (Zolg et al., 1989). Resistant P. falciparum possessed mutation in the DHFR gene at codon 108, the majority changing from Ser/Thr to Asn, Cys59 to Arg, and Phe223 to Ser (Reeder et al., 1996). Point mutation and gene amplification gave an important resistance mechanism to parasites. Isolated drug-resistant strains have been believed to be induced by continuous chemotherapy until now. But, with these results, the stability of DHFR gene was very strong to drug exposure, even though the DNA contents are in asexual stage (IN) that is expected to be much higher possibility of mutation. It was suggested that the mutated isolations might be caused by adaptation to drug resistance after random spontaneous mutation to any other environmental factors in addition to continuous exposure to drug only.
Notes
This study was supported by the research grant (No. 961-0716-101-1) from the Korean Science and Engineering Foundation (1996).