Microbiome of Haemaphysalis longicornis Tick in Korea
Article information

Abstract
Ticks can transmit pathogenic bacteria, protozoa, and viruses to humans and animals. In this study, we investigated the microbiomes of Haemaphysalis longicornis according to sex and life stages. The Shannon index was significantly higher for nymphs than adult ticks. Principal coordinates analysis showed that the microbiome composition of female adult and male adult ticks were different. Notably, Coxiella-like bacterium (AB001519), known as a tick symbiont, was found in all nymphs and female adult ticks, but only one out of 4 male adult ticks had Coxiella-like bacterium (AB001519). In addition, Rickettsia rickettsii, Coxiella burnetii, and Anaplasma bovis were detected in this study.
INTRODUCTION
Ticks can transmit pathogenic bacteria, protozoa, viruses that cause great suffering and potentially fatal diseases in humans worldwide [1]. Ticks also cause considerable losses to the livestock industry by causing irritation and blood loss to their hosts, thus decreasing the leather quality and at the same time acting as vectors of many pathogens [2]. Haemaphysalis longicornis has been reported in Australia, New Zealand, New Caledonia, Fiji, Japan, Korea, Northeastern China, USSR, and USA [3–6].
The bacterial pathogens transmitted by Haemaphysalis spp. are Rickettsia spp., Ehrlichia spp., Anaplasma spp., Francisella spp., Coxiella spp., and Borrelia spp. which cause the following diseases, respectively: Rocky Mountain spotted fever, Siberian or North Asian typhus, Japanese spotted fever, and Australian spotted fever; human monocytic ehrlichiosis and canine ehrlichiosis; human granulocytic anaplasmosis, and bovine anaplasmosis; tularemia; Q fever; and Lyme disease and tick-borne relapsing fever [7]. The parasites transmitted by Haemaphysalis spp. are Theileria spp., Babesia spp., and Hepatozoon spp. which likewise cause the following diseases, respectively: tropical theileriosis and sheep theilerosis; cattle, dog, and sheep babesiosis; and hepatozoonosis [7].
Despite the importance of H. longicornis as an important pathogen vector, little is known regarding its microbiome. In this study, we employed high-throughput sequencing of the V3–V4 hypervariable regions of the 16S rRNA gene to investigate the bacterial abundance and diversity between the nymph stage and the adult developmental stages of H. longicornis to evaluate the changes in the bacterial abundance and the maintenance of the bacterial community throughout the life cycle of the tick.
MATERIALS AND METHODS
Tick collection and identification
Ticks were collected within a radius of 50 m from vegetation (geographical location: 37.323366/129.233736) by flagging in Samcheok, Gangwon-do Province, Korea, in June 2020. Species identification of the collected ticks was performed by examination under a dissecting microscope, according to Yamaguti et al. [8]. Among the 140 H. longicornis obtained, 11 nymphs, 3 female adults, and 4 male adults were randomly selected for microbiome study.
DNA extraction
The surface of each tick was sterilized using alcohol before DNA extraction. DNA was extracted from individual ticks using a NucleoSpin DNA Insect kit (Macherey–Nagel, Düren, Germany) according to the manufacturer’s instructions.
High-throughput sequencing of 16S rRNA gene amplicons
The V3–V4 region of the 16S rRNA gene was amplified by PCR using the following bacterial universal primer pair: forward primer, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′ and reverse primer, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′ [9]. A limited-cycle amplification step was performed to add multiplexing indices and Illumina sequencing adapters. The libraries were normalized, pooled, and sequenced using the MiSeq platform (600 cycles, Illumina MiSeq V3 cartridge; Illumina, San Diego, California, USA) according to the manufacturer’s instructions.
Bioinformatics and statistics
Bioinformatic analyses were performed as described by Kim et al. [9]. Raw reads were processed through a quality check, and low-quality (Q <25) reads were filtered using Trimmomatic 0.32 [10]. Paired-end sequence data were then merged using PandaSeq [11]. Primers were trimmed using the ChunLab in-house program (ChunLab, Inc., Seoul, Korea), applying a similarity cut-off of 0.8. Sequences were denoised using the Mothur pre-clustering program, which merges sequences and extracts unique sequences, allowing up to 2 differences between sequences [12]. The EzBioCloud database [13] was used for taxonomic assignment using BLAST 2.2.22 [14], and pairwise alignments were generated to calculate similarity [15]. The UCHIME algorithm and non-chimeric 16S rRNA database from EzBioCloud were used to detect chimeric sequences for reads with a best hit similarity rate of <97% [16]. Sequence data were then clustered using CD-Hit and UCLUST [17,18]. All of the abovementioned analyses were performed with EzBioCloud, a commercially available ChunLab bioinformatics cloud platform for microbiome research (https://www.ezbiocloud.net/). The reads were normalized to 10,000 to perform the analyses. We computed the Shannon index [19], unweighted pair group method with arithmetic mean (UPGMA) clustering [20], and principal coordinates analysis (PCoA) [21] based on the generalized UniFrac distance [22]. We used the Kruskal–Wallis test to test for differences in the number of operational taxonomic units (OTUs) and used the Shannon index to compare microbiome diversity between the 2 age groups. We used linear discriminant analysis effect size (LEfSe) analysis to identify significantly different taxa [23]. Swarm plot was created by the beeswarm package in R software (version 4.0.5).
RESULTS
For H. longicornis nymph, female adult, and male adult ticks, the average reads assigned to bacteria were 35,622, 59,365, and 51,859, respectively (Supplementary Table S1). The number of OTUs, which reflects species richness, was not significantly different between the groups (Fig. 1A, B; Supplementary Table S1). The Shannon index, which reflects species diversity, was significantly higher for nymphs (n=11) than the adult ticks (both males and females, n=7; P=0.026; Fig. 1C). However, it did not differ significantly among the 3 groups, and the average Shannon indices were 1.55, 0.83, and 0.78 for nymphs (n=11), female adults (n=3), and male adult (n=4) ticks, respectively (Fig. 1D; Supplementary Table S1).
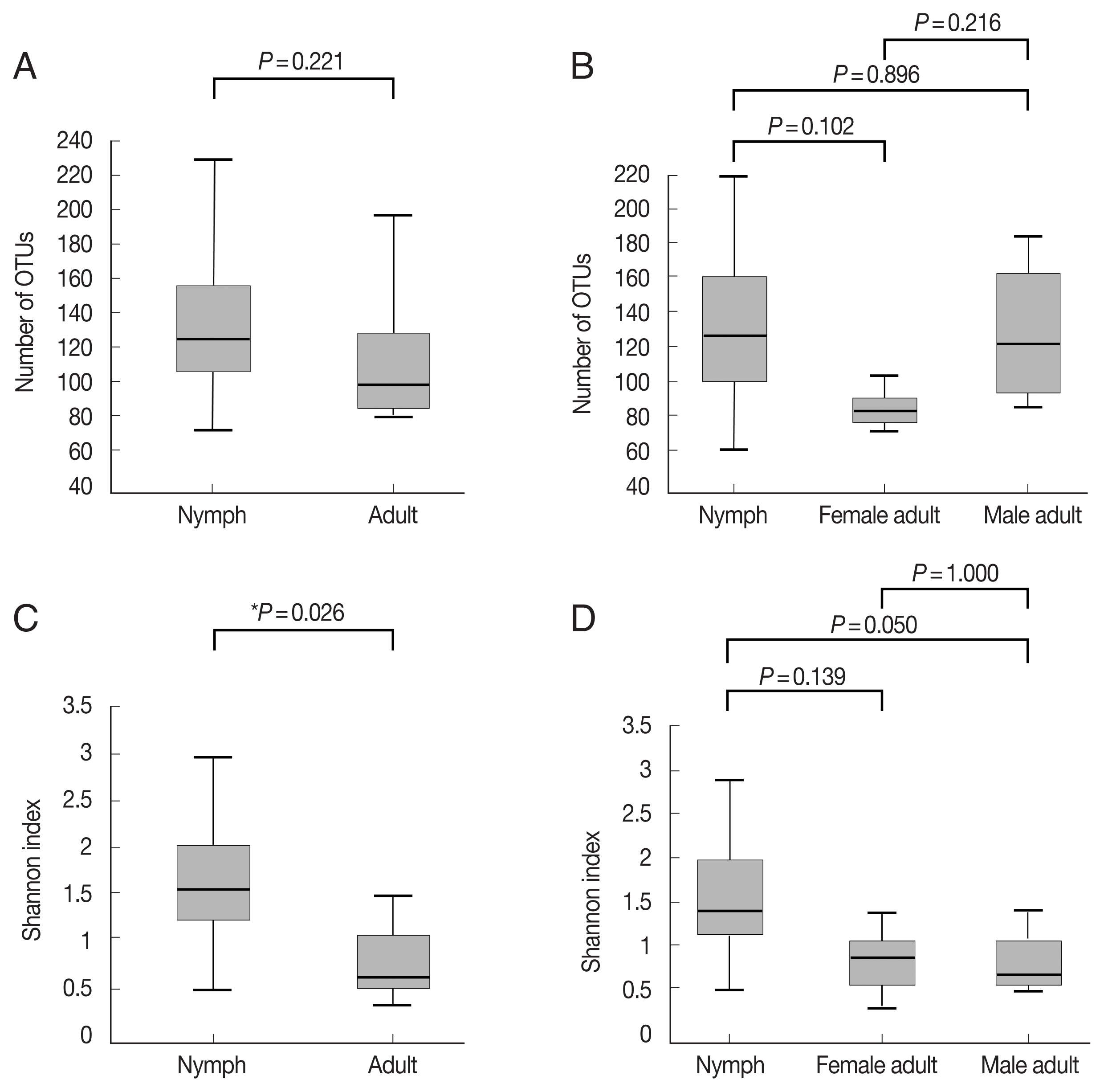
Alpha diversities of the microbiomes of Haemaphysalis longicornis. (A) The number of OTUs (species richness) in nymph (n=11) and adult tick (n=7) groups. (B) The number of OTUs in nymph (n=11), female adult tick (n=3), and male adult tick (n=4) groups. (C) The Shannon index in nymph and adult tick groups. (D) The Shannon index in nymph, female adult tick, and male adult tick groups. The boxplots indicate the minimum, first quartile, second quartile (median), third quartile, and maximum values.
Beta diversity was analyzed to show the difference in bacterial composition between the samples. First, the PCoA result showed that the samples of the male adult group and those of the female adult group clustered well (Fig. 2A), suggesting that the bacterial composition of the 2 groups was different. In contrast, nymph samples were relatively scattered in the plot versus the other groups. Similarly, in UPGMA clustering, the male adult and female adult samples were clustered closely among their groups than the nymph samples.
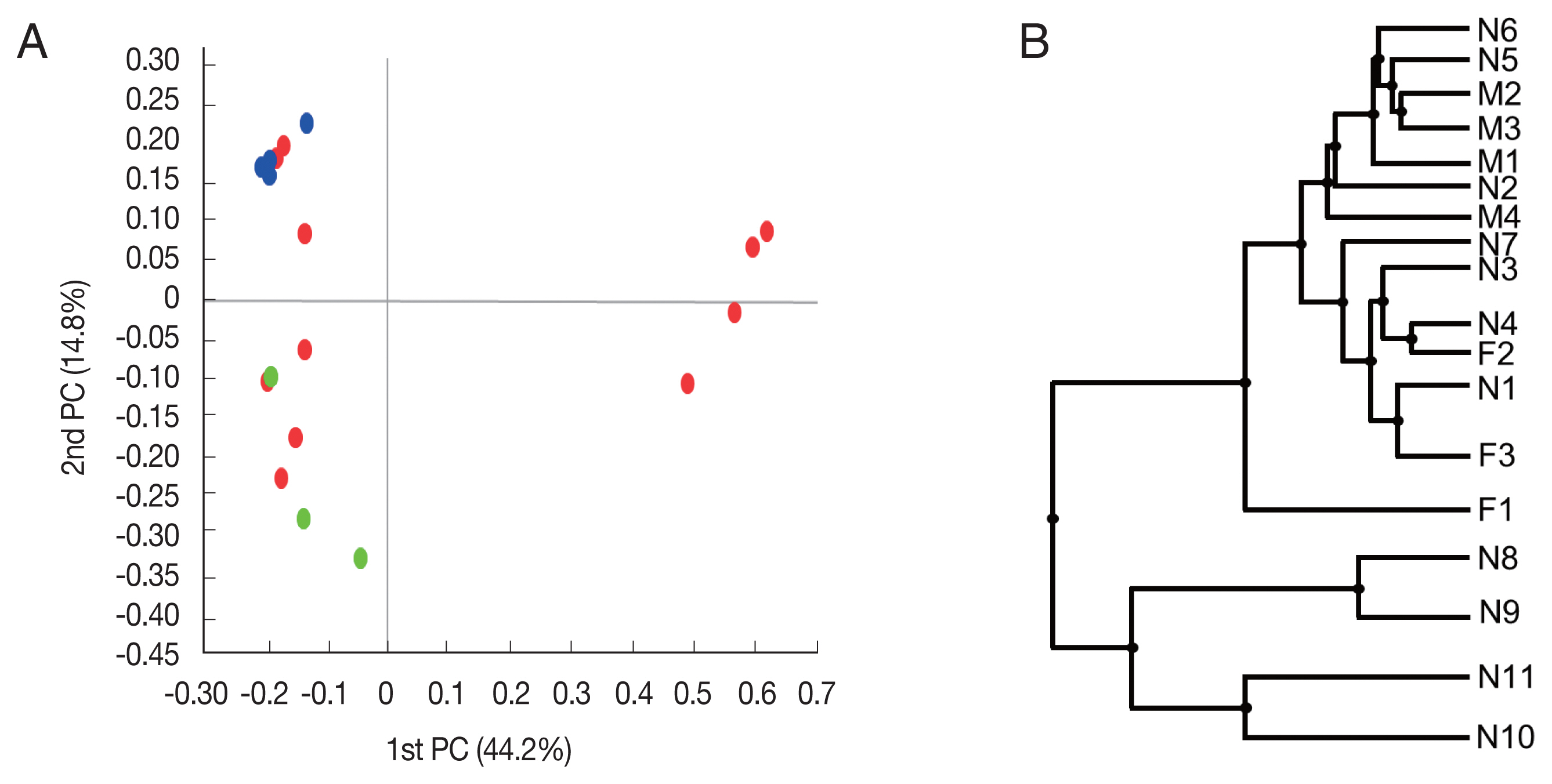
Beta diversities of the microbiomes of Haemaphysalis longicornis. (A) Principal coordinates (PC) depicting differences in the taxonomic compositions of the bacterial communities. Nymph (red), female (green), and male adults (blue). (B) Unweighted pair group method with arithmetic mean (UPGMA) clustering. N1–N11 represent nymphs, F1–F3 represent female adult ticks, and M1–M4 represent male adult ticks.
In the composition, the most predominant bacterial species was Pseudomonas fulva, which was found in all ticks evaluated in this study with an average relative abundance of 48.9% (Fig. 3A, B). The second most abundant species was Coxiella-like bacterium (AB001519), a known tick endosymbiont [24], with an average relative abundance of 20.7% (Fig. 3A). Coxiella-like bacterium (AB001519) was found in all nymphs and female adult ticks although some nymph samples showed very few reads for this species (Fig. 3B; Supplementary Table S2). In contrast, among the 4 male adult tick samples, Coxiella-like bacterium (AB001519) was found in only one sample with 0.7% relative abundance (Fig. 3B). Relative abundance of Corynebacterium spp., including C. falsenii, C. xerosis, and C. resistens was more than 40% in 4 nymphs (N8, N9, N10, and N11) but it was not detected in other samples (Fig. 3B).
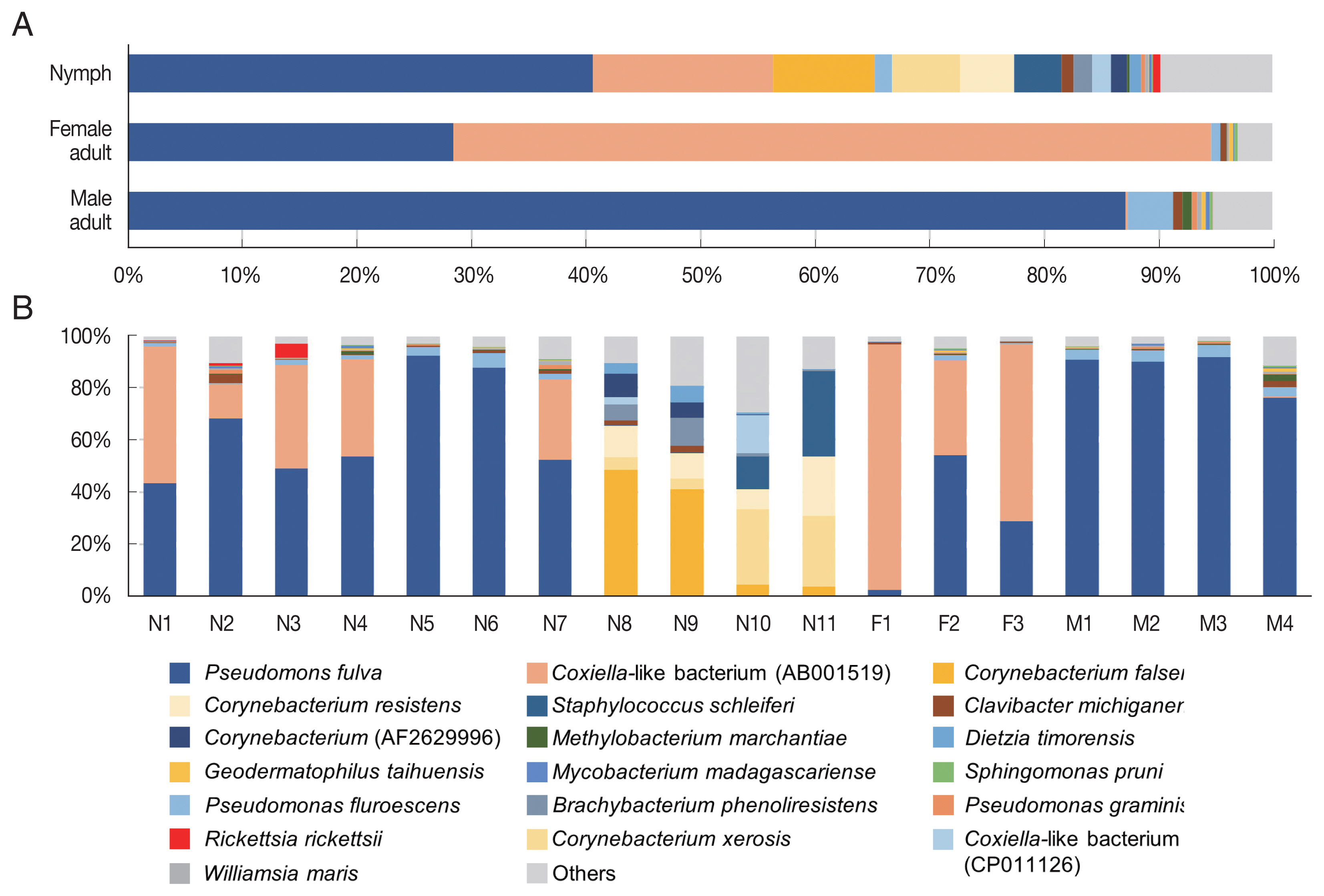
Microbiome composition at species level in Haemaphysalis longicornis. (A) Average microbiome composition in the ticks. (B) Individual microbiome composition of the nymph (N1–N11), female (F1–F3), and male (M1–M4) ticks.
To identify significant differences in bacterial abundance between the nymph, female adult, and female adult ticks, we performed LEfSe analysis. The LDA scores for Coxiella-like bacterium (AB001519) was 4.88 and its average relative abundance was 66.4% in female adult ticks, 15.7% in nymphs, and 0.2% in male adult ticks (Table 1; Fig. 4A). The LDA score for P. fulva was 4.79 and its relative abundance was the highest in male adult ticks (86.9%, Fig. 4B). The LDA score for P. fluorescens was 3.51 and its relative abundance was the highest in male adult ticks (4.0%, Fig. 4C).
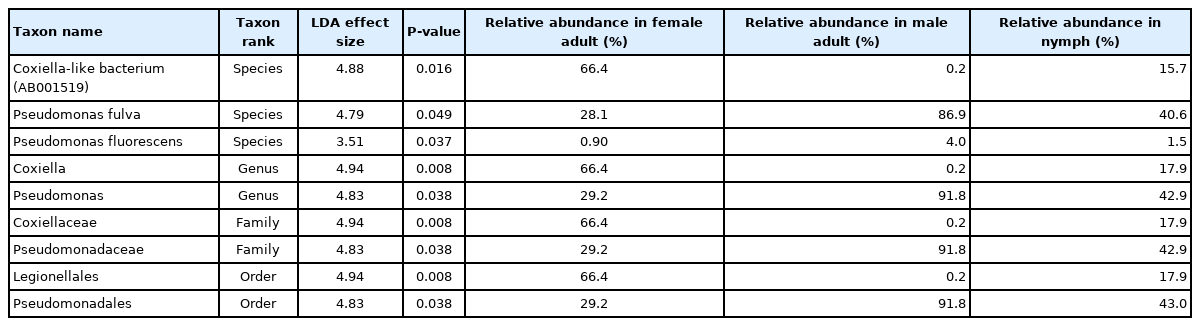
Linear discriminant effect size analysis on differentially abundant bacterial taxa between Haemaphysalis longicornis nymph, female and male ticks
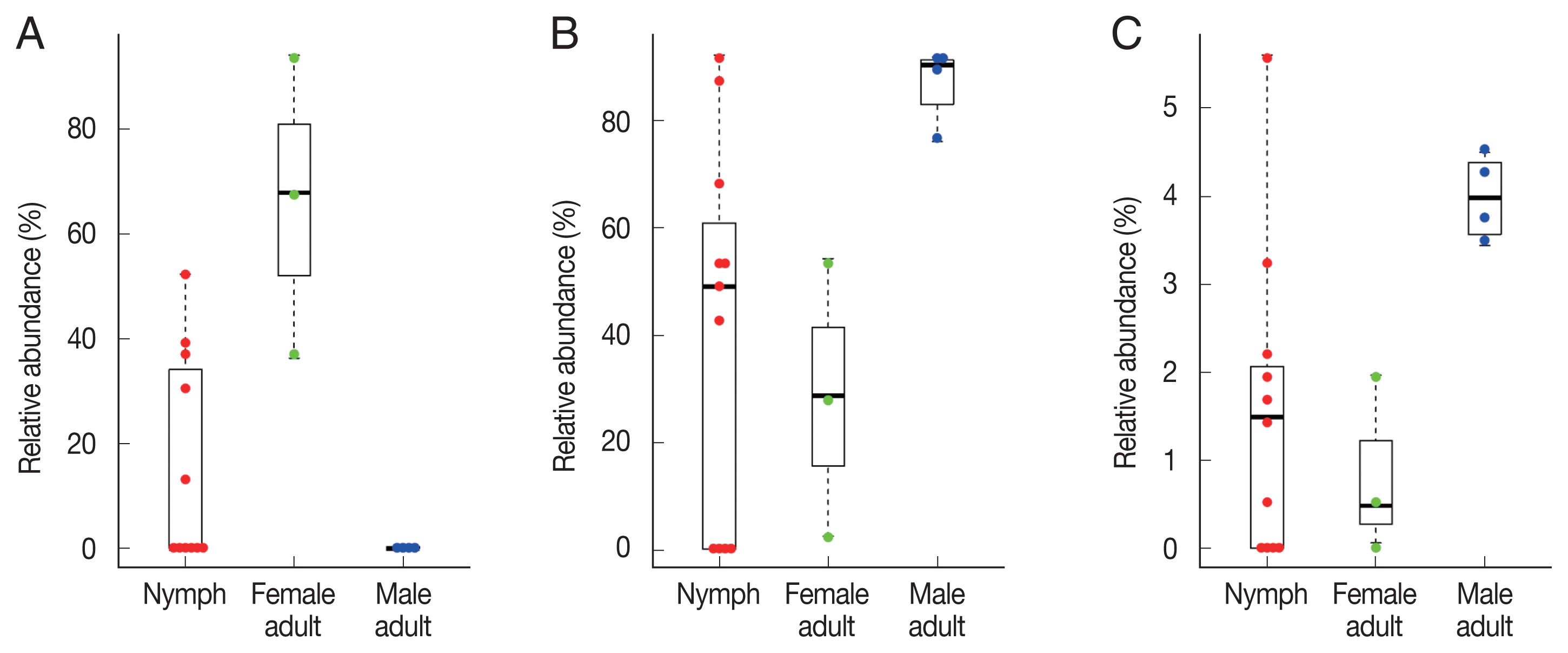
Swarm plots of the relative abundance in Haemaphysalis longicornis. (A) Coxiella-like bacterium (AB001519). (B) Pseudomonas fulva. (C) Pseudomonas fluorescens.
Known pathogenic species were also identified. Rickettsia rickettsii was detected in 3 nymphs and 3 male adult ticks and Coxiella burnetii was detected in 4 nymphs (Supplementary Table S2). In addition, Anaplasma bovis, which infects ruminants, was found in one nymph (Supplementary Table S2). Ehrlichia spp., Francisella spp., and Borrelia spp. were not detected in this study.
DISCUSSION
H. longicornis is the most common tick species in the grass in Korea. According to previous studies, out of 7,973 ticks collected from farms in Korea, 7,758 (97.3%) were identified as H. longicornis [25,26]. H. longicornis is also known as a transmitter of several pathogens such as Rickettsia spp., Anaplasmataceae, Borrelia spp., Babesia spp., Francisella spp., Bratonella spp., Coxiella spp., and severe fever with thrombocytopenia syndrome virus [27].
In this study, we investigated the differences in the microbiomes of H. longicornis nymphs, female adults, and male adults. The Shannon index was significantly higher for nymphs than the adult ticks (Fig. 1C). This suggests that nymphs harbor a more diverse bacterial community compared to adult ticks. In 6 species of ticks, including Haemaphysalis leporispalustris, microbiome richness and diversity were highest in the larval stage and eventually decreased in subsequent life stages; this result is in concordance with our findings [28]. Life-stage related differences in the microbiome of H. longicornis indicate that the tick loses most of the bacteria present in the early developmental stages and then reestablishes the bacterial community after molting [29,30].
Coxiella spp. is known as an endosymbiont essential for the survival and reproduction of hard ticks [26,31]. Coxiella-like bacteria are a large group of yet-to-be-isolated and characterized bacteria phylogenetically close to the causative agent of Q fever, C. burnetii [32]. These Coxiella-like bacteria are maternally inherited endosymbionts, highly prevalent in tick populations, and engaged in mutualistic interactions with their arthropod hosts [33]. In our study, the relative abundance of Coxiella-like bacterium (AB001519) was significantly different among nymphs, female adult, and male adult ticks. An average of 66.4% of female adult ticks and all nymphs and female adult ticks were found to have Coxiella-like bacterium (AB 001519). In contrast, only one tick out of 4 male adult ticks had Coxiella-like bacterium (AB001519), and its relative abundance was only 0.7%. Coxiella-like bacterium (AB001519) may be playing an important role in the development and reproduction of H. longicornis especially for female adult and during the nymph stage. Since Coxiella-like bacterium (AB001519) is almost eliminated in male adult ticks, there may be no role for this bacterium in male adult ticks; likewise, this bacterium may be transmitted vertically from mother tick to offspring. In addition, at least one strain of Pseudomonas was identified in each tick in this study. Pseudomonas spp. were frequently detected in ticks in other studies, indicating that it may have a symbiotic association with ticks [34,35].
Among the 18 tick samples in this study, R. rickettsii was found in 6, C. burnetii in 4, and A. bovis in 1, although further investigation is necessary to determine the pathogenicity of the isolated strains. R. rickettsii is the causative agent of Rocky Mountain spotted fever, one of the most virulent human infections [36]. Using molecular assays, several spotted fever-causing Rickettsia spp., including R. japonica, R. conorii, R. akari, R. felis, R. rickettsii and unclassified Rickettsia spp., have been detected in humans, ticks, and mites [37–39].
C. burnetii is an obligate intracellular bacterial pathogen causing Q fever that is manifested by pneumonia, fever, and granulomatous hepatitis. The reservoir of C. burnetii includes mammals, birds, and arthropods such as ticks [40]. Domestic animals represent the most frequent source of human C. burnetii infection [40]. The number of human Q fever cases in Korea has been rapidly increasing since 2015 [41]. Several studies have investigated C. burnetii infection in dairy cattle in Korea [42].
A. bovis can be transmitted by ticks to cattle, sheep, goats, and other domestic ruminants [43]. A. bovis causes fever, anemia, drowsiness, convulsions, weight loss, and enlargement of lymph nodes in cattle [44]. Recently, A. bovis was also detected in Korean spotted deer (Cervus nippon) [45], Korean water deer (Hydropotes inermis) [46], and H. longicornis in Korea [47,48].
A limitation of this study was that we did not investigate the microbiome of H. longicornis in its larval stage. A single larva is not sufficient for the effective extraction of DNA, which is required to further perform high-throughput sequencing. However, the strength of this study was that we were able to investigate the microbiome of the individual ticks without pooling their DNA samples. In addition, ticks were collected from a relatively small location and at a single time point. Further similar studies on a larger number of ticks collected from various locations and at multiple time points are required.
In conclusion, in this metagenomics study on H. longicornis, different microbiome patterns were found among nymphs, female adult ticks, and male adult ticks. Coxiella-like bacterium (AB001519) was found in all nymphs and female adult ticks, but rarely in male adult ticks. In addition, several potential pathogens were found such as R. rickettsii, C. burnetii, and A. bovis.
Supplementary Information
ACKNOWLEDGMENTS
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean Government (MEST; numbers NRF-2019R1A2B5B01069843, 2020R1I1A2074562).
Raw sequence data are available in NCBI GenBank under Bioproject PRJNA733831.
Notes
The authors have no competing interests to be declared.