Echinostomes are intestinal flukes that are widespread in Southeast Asia. Four species are prevalent in Korea; Echinostoma hortense, Echinostoma cinetorchis, Echinochasmus japonicus, and Acanthoparyphium tyosenense. Infections by echinostomes are associated with more severe symptoms than those due to other intestinal flukes, because they are larger than other flukes and adults have head crowns and collar spines that can cause serious stomach damage [1].
This fluke has a head crown with 37–38 collar spines, including 5 spines on both ends. It also has testes that are not constant in number and are not fixed in a single location [2]. Flukes were studied mainly by Japanese researchers until 1960, with a focus on human infections [3–5], intermediate hosts, reservoir hosts, and biological and epidemiological characteristics [1]. As a result of these studies, large shellfish, frogs, tadpoles, and loaches were found to act as the first intermediate hosts [1,6,7]. The fluke has been found to be widespread in Rattus norvegicus, which lived in inland of Korea during nationwide surveys on infection of reservoir hosts with these flukes [8].
Recent researches on parasites have focused on developing vaccines and chemotherapeutic drugs by elucidating the pathological and biochemical characteristics of specific molecules, especially proteinases and oligonucleotide targeting. However, there was been very little biochemical and molecular biological research on E. cinetorchis, as the majority of studies on this species have focused on the life cycle and intermediate host exploration.
Therefore, we cloned and examined the characteristics of an endoribonuclease of E. cinetorchis.
Endoribonuclease can cut single-stranded and double-stranded RNA. The proteins include RNase III, RNase A, RNase T1, RNase H, RNase P, and RNA-induced silencing complex. Turnover of mRNA is very important for gene regulation. Exonucleases are known to be involved in mRNA degradation that controls gene expression and has 5′-3′ exonucleolytic and 3′-5′ exonucleolytic activity. However, little is known about endoribonucleases that degrade RNA at internal sequences [9]. Recently, RNase H (EC 3.1.26.4) of Leishmania was investigated for understanding the function of it in this parasitic organism, which will lead to more effective and optimized chemotherapeutic drug-targeting strategies against the harmful afflictions it causes [10]. Therefore, we analyzed the DNA sequence of endoribonuclease from E. cinetorchis and characterized a recombinant endoribonuclease protein to enhance developing a new antisense oligonucleotide targeting to damage the parasites. In addition, we developed monoclonal antibodies to determine the location of endoribonuclease in adult E. cinetorchis and to obtain valuable information related to pathology and diagnosis.
Naturally infected Segmentina (polypylis) hemisphaerula were collected in Sinchang-myon, Asan-si, Chungnam Province, and then metacercariae were isolated. Five Sprague-Dawley rats (body weight: 120 g) were infected with 50 metacercariae using a tuberculin syringe and maintained at 20°C in 50–55% humidity and a 12 hr-light/12 hr-dark cycle for 4 weeks. The presence of E. cinetorchis eggs were confirmed by microscopic examinations. Adult E. cinetorchis worms were isolated from the small intestine of a rat by dissection. To amplify the E. cinetorchis endoribonuclease, degenerative primers described below were designed based on consensus sequences of Schistosoma haematobium (NCBI GenBank no. KGB32409.1), Schistosoma japonicum (AAW25442.1), Clonorchis sinensis (GAA56298.1), and Schistosoma mansoni (CCD59795.1). RT-PCR for amplification of the E. cinetorchis endoribonuclease was performed with the mRNA prepared above. The amplified product was inserted into a pGEM-T Easy vector (Promega, Madison, Wisconsin, USA). The sequence did not contain 5′ and 3′ ends; therefore, 3′-RACE was performed with a 3′-Full RACE Core Set (Takara Bio Inc., Otsu, Shiga, Japan) and specially designed primers: sense primer, 5′-YTN YTN GAY AAY TAY-3′ (15 mers), and anti-sense primer, 5′-CTG ATC TAG AGG TAC CGG ATCC-3′ (21 mers) (Y=T or C, N=A or T or C or G). 5′-RACE was performed with a 5′-Full RACE Core Set (Takara Bio Inc.) and sense primer 5′-ATG ATT GCG TGG CCA ACA GTG-3′ (21 mers) and anti-sense primer 5′-ATC GTT GTC AGC GCT CCA CC-3′ (20 mers).
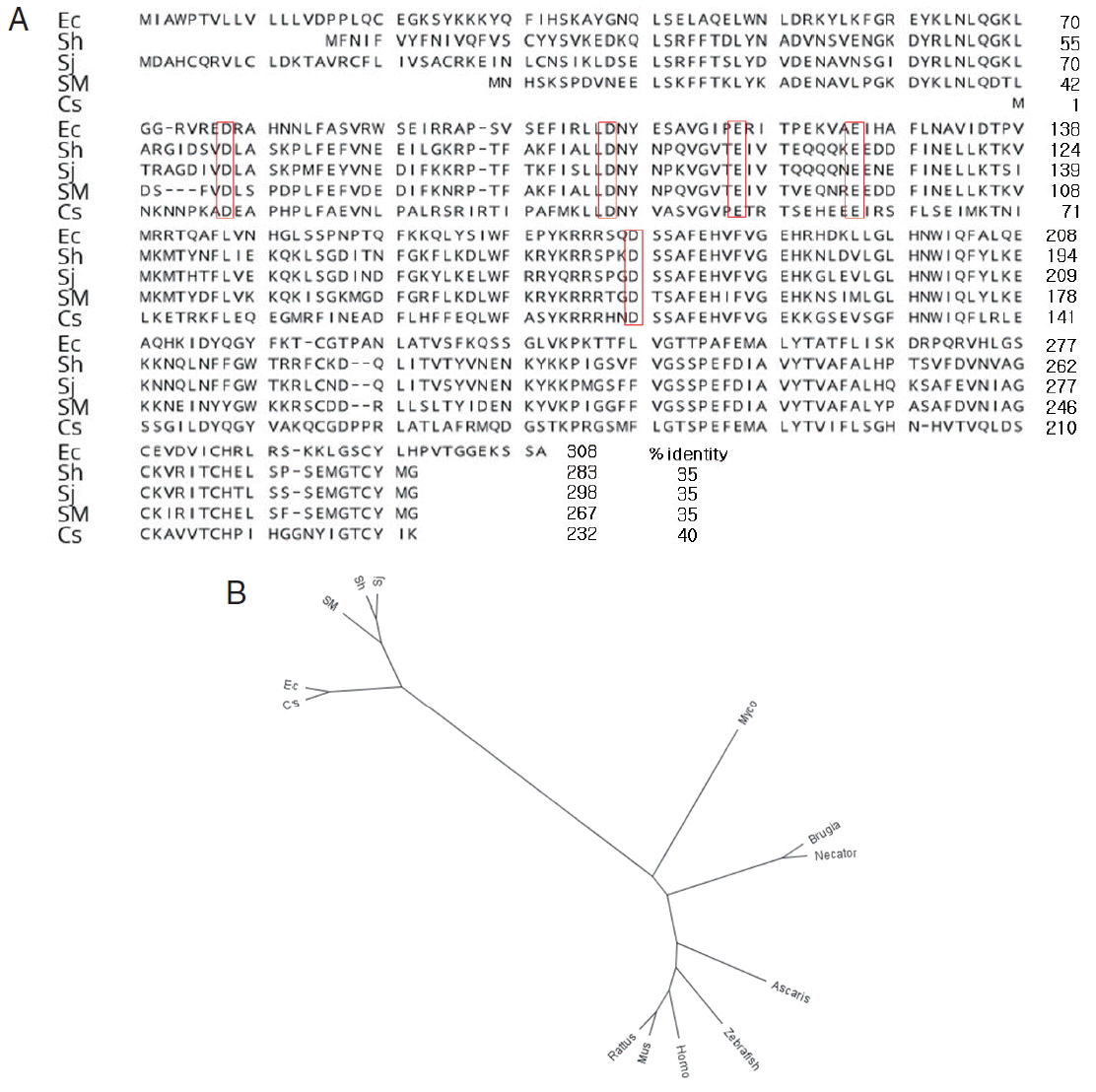
It was shown that the cDNA sequence E. cinetorchis endoribonuclease consists of 927 nucleotides, which encoded 308 amino acids, as determined by the DNASIS program. It is known that RNases H have an active site centered on a conserved motif composed of aspartate (D) and glutamate (E) residues, DEDD motif. Interestingly, we found putative DDEED motif instead of DEDD motif in parasites (Fig. 1A) [12].
The amino acid sequences showed 35% similarity with that of S. haematobium, 35% with that of S. japonicum, 35% with that of S. mansoni, and 40% with that of C. sinensis, 6% with that of Mycobacterium tuberculosis (AGQ35548.1), 7% with that of Rattus norvegicus (AAH91209.1), 8% with that of Mus musculus (AAH19411.1), 8% with that of Homo sapiens (AAH02973.1), 5% with that of Necator americanus (ETN82101.1), 6% with that of Brugia malayi (XP_001895788.1), 5% with that of Ascaris suum (ERG86215.1), and 5% with that of Danio rerio (AAH76457.1), whereas endoribonucleases in S. haematobium, S. japonicum, and S. mansoni showed greater than 70% sequence similarity. Based on the low similarity of the E. cinetorchis ribonuclease with those of other parasites, it might have a unique character than other genes (Fig. 1A, B). Serine proteases, cysteine proteinases, and RNases of protozoans and helminths have been studies extensively. However, little is known about endoribonuclease in S. haematobium, S. japonicum, and S. mansoni. Therefore, we provided the first report on the molecular biological characteristics of the endoribonuclease in E. cinetorchis, including the full cDNA sequence.
To express a recombinant endoribonuclease protein in E. coli, an EcoRI (GAATTC) restriction site was added to the sense primer, 5′-GAATTC ATGATTGCGTGGCCAACAGTG-3′ (27 mers), and a XhoI (CTCGAG) sequence was added to the anti-sense primer, 5′-CTCGAG ATCGTTGTCAGCGCTCCACC-3′ (26 mers). The PCR product was inserted into a pGEM-T Easy vector (Promega) and transformed into E. coli DH5α for gene amplification. After confirming the DNA sequence of the coding region in pGEX-4T-1 (GST-tagged) expression vector, we obtained a transformed recombinant protein expressed clone, named E. coli BL21(DE3) plysS[pEndoEx]. The GST-tagged endoribonuclease protein was purified with GST-binding agarose resin (ELPIS-Biotech Inc., Daejeon, Korea) according to the manufacturer’s manual. A soluble protein was successfully obtained, as confirmed by GST-specific antibody in a western blot analysis. The GST tag was removed by treatment with 10–20 units thrombin (Fig. 2, lane 3) and cleaned with a Sephacyl S-200 High Resolution Gel (Fig. 2, lane 4). Based on results of the SDS-PAGE, the GST-free recombinant endoribonuclease protein showed a molecular weight of approximately 34 kDa (Fig. 2, lane 4). Several expression vectors, pET29a (His-tagged), pQE30 (His-tagged), and pBAD (His-tagged), and Escherichia coli M15 cell and BL21 (DE3) plysS as competent cells, were used in several combinations to express the cloned endoribonuclease gene; however, these attempts failed (data not shown). Even in several combinations, finding the best conditions to obtain the soluble protein was challenging. When E. coli BL21(DE3) plysS[pEndoEx] cells were cultured at 37°C with 0.5–1.0 mM IPTG, a large quantity of insoluble protein (inclusion bodies) was expressed. Even in culture at 25°C, inclusion bodies were observed (data not shown). Interestingly, small amounts of soluble proteins were obtained when the cells were cultured at 16°C for 24–48 hr.
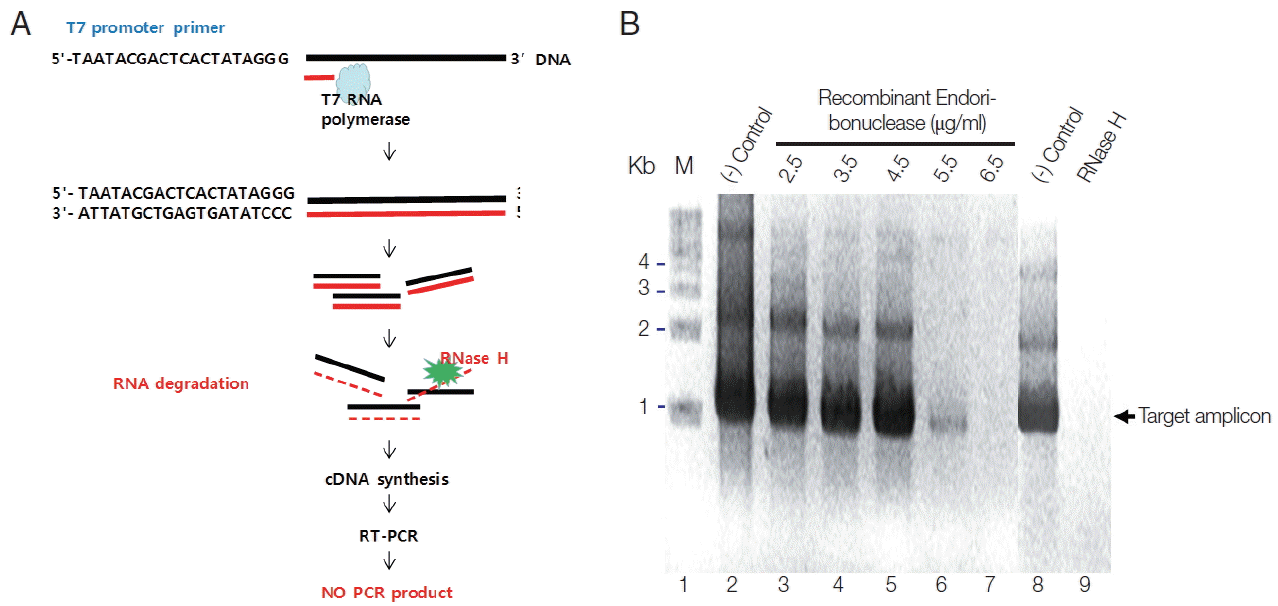
To verify the enzymatic activity of recombinant endoribonuclease, Parniak’s method was modified as described below [11]. To do this, first of all, we synthesized RNA from T7 promoter conjugated full cDNA of endonuclease using T7 RNA polymerase (Fig. 3A). Then, we treated them with various concentrations of endonuclease or RNase H to allowed RNA degradation. To see the various concentrations of amplified DNA according to their degraded RNA by the role of enzymes, RT-PCR was done. One unit of endoribonuclease was defined as not produce the PCR product according to the character of RNase H. When adding purified recombinant endoribonuclease protein at different concentrations (2.5, 3.5, 4.5, 5.5, and 6.5 μg) into the plasmid DNA, pEndo (the template) and the amplified PCR products showed the same amount according to the concentration of endoribonuclease (Fig. 3B, control lanes). Because the DNA was not the endoribonuclease substrate, it did not hydrolyze the DNA. However, by adding the DNA-RNA hybrid as a template for PCR, the amplified PCR products were reduced according to the endoribonuclease concentration. At an endoribonuclease concentration of 6.5 μg, PCR product was not obtained (Fig. 3B, lane 7). Therefore, 1 unit of endoribonuclease was defined as 6.5 μg. When adding 1 unit of RNase H (Takara) to the PCR reaction, no PCR product was obtained (Fig. 3B, lane 9), similarly to the result with the recombinant endoribonuclease protein (Fig. 3B, lane 7). Based on these results, purified recombinant endoribonuclease protein was considered an RNase H. Therefore, modified enzymatic assay for RNase H activity may be useful to examine these kinds of enzymes based on PCR.
Generally, endoribonucleases contribute to mRNA stability, but the function of this endoribonuclease in cells is little known; for example, it has not yet been shown how this enzyme cuts the specific RNA site. Exceptionally, how Dicer and AGO2 recognize the substrates well? The intracellular location of endoribonucleases is very important to understanding the cleavage specificity of an endoribonuclease for dinucleotide sequences [9]. Biologically, RNase H appears to be involved in DNA replication and repair and possibly in transcription. By targeting antisense phosphorothioate oligodeoxynucleotides (ODN) to the essential mRNAs in Leishmania, the RNA–DNA hybrid-specific cleavage feature of RNase H can be manipulated to operate in the host’s advantage. The ablation of gene expression in the parasite can prevent its proliferation or lead to its death. Different classes of RNase H have been detected and characterized in several organisms ranging from bacteria to retroviruses to mammals, and it is interesting that they all share some homology at different levels. Theoretically, co-treatment of Leishmania with a possible activator of RNase H and an antisense oligonucleotide should accentuate the efficacy of the ODN and bring plausible chemotherapy treatment for leishmaniasis [10]. Therefore, our finding also contributes to developing new strategies based on antisense ODN to kill the parasites, including E. cinetorchis. However, we do not know yet why this protein located in the muscle layer only (data not shown); it may be expressed as essential proteins for parasite survival in this area. Therefore, it should be investigated in more details in the near future.