Mitochondrial Genome Sequences of Spirometra erinaceieuropaei and S. decipiens (Cestoidea: Diphyllobothriidae)
Article information
Abstract
The present study was performed to compare the mitochondrial genomes between 2 Spirometra tapeworms, Spirometra erinaceieuropaei and Spirometra decipiens (Cestoidea: Diphyllobothriidae), which larval stages are important etiological agents of sparganosis in humans. For each species, the full mitochondrial genome was amplified in 8 overlapping fragments using total genomic DNA purified from a single worm as the template. The mitochondrial genomes were 13,643 bp (S. erinaceieuropaei) and 13,641 bp (S. decipiens) in length and contained 36 genes; 12 protein-coding genes, 2 ribosomal RNA (rRNA, small and large subunits), and 22 transfer RNAs (tRNAs). The 12 protein-coding genes constituted 10,083 bp (S. erinaceieuropaei) and 10,086 bp (S. decipiens) of their respective mitochondrial genomes. The tRNA genes, ranging in length from 56 to 70 bp, were identified based on putative secondary structures such as the typical cloverleaf shape. A total of 23 intergenic sequences, varying from 1 to 204 bp in size, were interspersed in S. erinaceieuropaei (total, 504 bp) and S. decipiens (total, 496 bp) mtDNA. The 12 protein-coding genes of S. erinaceieuropaei and S. decipiens differed by 12.4%, whereas the overall difference in mtDNA sequence between S. erinaceieuropaei and S. decipiens was 12.9%. Thus, from the standpoint of the mitochondrial genome, S. decipiens represents a valid species that can be distinguished from S. erinaceieuropaei.
INTRODUCTION
Spirometra erinaceieuropaei and S. decipiens are pseudophyllidean cestodes. In humans, these worms can induce sparganosis, and their medical importance is well- established. Humans can be infected by procercoid larvae in cyclops (the first intermediate host) and plerocercoid larvae in reptiles or amphibians (paratenic hosts). The genus Spirometra was established by Mueller [1], but this name had been used earlier by Faust et al. [2], who proposed dividing the genus into 2 subgenera, Diphyllobothrium and Spirometra.
S. erinaceieuropaei was first reported by Rudolphi as Dubium erinaceieuropaei from hedgehogs (Erinaceus europaeus) in 1819. Diesing in 1853 recognized this larva as a sparganum and named it Sparganum erinaceieuropaei. Molin in 1895 renamed it as Sparganum lanceolatum, which is regarded as a synonym. These names refer to the larval stage of spirometrid species. Based on morphological characteristics described by Diesing [2], S. decipiens is synonymous with 4 species; Bothriocephalus felis Creplin, 1825, Bothriocephalus maculatus Leukart, 1848, Dibothrium decipiens, and Bothriocephalus decipiens Diesing, 1850. In 1929, Faust, Campbell and Kellogg reviewed the morphological characteristics of S. erinaceieuropaei and S. decipiens by monitoring the experimental development of adults from spargana in humans and other vertebrates.
Currently, the species classification in the genus Spirometra is controversial. Despite the availability of reliable diagnostic morphological characters for species identification, the specieslevel taxonomy of Spirometra spp. remains obscure. Therefore, it is necessary to devise a reliable taxonomic criterion based on morphological characteristics in combination with molecular analysis of mitochondrial DNA sequences. Mitochondrial DNA sequencing is now considered a useful molecular tool for inferences of evolutionary analysis, phylogenetic reconstruction, taxonomic identification, biogeography, population genetics, and epidemiological investigation [3,4]. The complete mitochondrial genomes of order Pseudophyllidea have been recently published for Diphyllobothrium latum [5], Diphyllobothrium nihonkaiense [6], and S. erinaceieuropaei [7].
Within the genus Spirometra, mitochondrial DNA sequences have also been used as genetic makers for identification and characterization of genetic variation of species [8,9]. The mtDNA cox1 gene sequences revealed that S. erinaceieuropaei and S. proliferum are distinct species [10]. More recently, the complete mitochondrial genome of S. erinaceieuropaei isolated from China was reported and compared to the mitochondrial genome of a Japanese isolate [7]. However, the speciation of the parasite may need to revise careful reconsideration based on morphological analyses of Spirometra tapeworms. The aim of this study was to provide information useful for classification of Spirometra spp., by the mitochondrial genome of S. erinaceieuropaei and S. decipiens and the mitochondrial genomes with those of other cestodes.
MATERIALS AND METHODS
Worm samples
Plerocercoid larvae of S. decipiens were collected from Rhabdophis tigrinus tigrinus, which were donated by the Association of Wildlife Protection, and then used to infect a cat, in April 2008. Plerocercoid larvae of S. erinaceieuropaei were collected from a 58-year-old woman, and then used to infect a dog in May 2009. Eight weeks after the infection, the cat and the dog were sacrificed, and adult worms were recovered from their intestines. Experimental animals were used following the ethical guidelines in commission of laboratory animals in Chungbuk National University (2008). The tapeworms were pressed and fixed in alcohol-formalin-acetic acid (AFA) for carmine staining. The following anatomical features of mature and gravid proglottids were observed, based on the morphological data of Faust et al. [2]; vaginal opening, uterus, uterine pore, cirrus, genital pore, testes, and vitellaria. Gravid proglottids were preserved at -70˚C in 70% ethanol until use. A single proglottid was chopped into small pieces, and total genomic DNA was extracted using the DNeasy tissue Kit (Qiagen, Valencia, California, USA).
PCR and DNA sequencing
For each species, the full mitochondrial genome was amplified in 8 overlapping fragments using total genomic DNA purified from a single worm as the template. The overlapping fragments of S. decipiens and S. erinaceieuropaei mtDNA were amplified using 16 oligonucleotide primers. The primers were used to amplify the cob-nad2 region (Spiro-cob-F1: TTT YCA YTC TTA TTT TAC YAC TAA GAA and Spiro-nad2-R1: AYC ACA CAT ACT CCC ARC TTG GGC TAC, 3.1 kbp), nad2-nad1 region (Spiro-nad2-F1: TTT GGG CST YKT GTT GYR TGT GTT ATT and Spiro-nad1-R1: CCA ACC RGC ACA CAA TAA AGC ATA ACT, 1.0 kbp), nad1-cox1 region (Spiro-nad1-F1: TAT GCY GAG TCG GAG AGG GAG TTG GTT and Spiro-cox1-R1: AAC MCC AAT AAT CAT AGT YAC AGA ACT, 2.1 kbp), cox1-rrnS region (Spiro-cox1-F1: GAC TGG TAA GTT AAT TTA AAC TGT and Spiro-12S-R1: CAT CTA ACC CAA CCG TAA CAT A, 2.5 kbp), rrnS-nad5 region (Spiro-12S-F1: GTA TTA ATA TTT AAG CTA AGT CTA TGT GCT and Spiro-nad5-R1: AAA CRC ACC AAG CAA TTT TAT TAC AGG TRG, 3.0 kbp) and nad5-cob region (Spiro-nad5-F1: CYA CCT GTA ATA AAA TTG CTT GGT GYG TTT and Spiro-cob-R1: YAA ACA AAC ATG AGC TGA AAA WAC ACG AAC, 2.5 kbp). PCR and DNA sequencing was performed as described previously [11].
Sequencing analysis
Sequences were assembled and aligned using Geneious 6.1.5 program (Biomatters Co., Auckland, New Zealand). The sequenced regions were identified using BLAST searches and compared with platyhelminth sequences in the GenBank database. Protein-coding genes were identified based on similarity of inferred amino acid sequences to those of other platyhelminth mtDNAs, as well as multiple comparisons with mitochondrial gene sequences in the GenBank database. The mitochondrial genetic code of platyhelminths was used to obtain putative translational products of the mitochondrial proteincoding sequences. Two ribosomal RNA genes (12S and 16S subunits) were determined by alignments with other known rRNA genes of platyhelminths. Twenty-two putative tRNA genes were identified using the tRNAscan-SE software [12] and anticodon sequences. The putative stem-loop structures of non-coding mitochondrial regions were inferred using the RNAdraw program [13].
Phylogenetic analysis was conducted using PAUP 4.0 [14]. The following mitochondrial genome sequences were used: S. erinaceieuropaei (KJ599680; this study), S. decipiens (KJ599679; this study), Diphyllobothrium latum (NC_008945), D. nihonkaiense (NC_009463), Echinococcus granulosus (NC_008075), E. multilocularis (NC_000928), Hymenolepis diminuta (NC_002767), Taenia solium (NC_004022), T. saginata (NC_009938), T. asiatica (NC_004826), and T. crassiceps (NC_002647). Phylogenetic trees were constructed using the maximum likelihood (ML), maximum parsimony (MP), neighbor joining (NJ), and Bayesian inference [15], with Fasciola hepatica (NC_002546) and Paragonimus westermani (NC_002354) as outgroups. Confidence values for tree branches were determined by bootstrap analyses with 1,000 replicates.
RESULTS
Gene content and organization
The mitochondrial genomes of Spirometra tapeworms were 13,643 bp (S. erinaceieuropaei) and 13,641 bp (S. decipiens) in length. The genomes each contained 36 genes, of which 12 were protein-coding genes, 2 were ribosomal RNA (rRNAs, small and large subunit), and 22 were transfer RNAs (tRNAs) (Fig. 1). As with other cestodes, both genomes lacked the atp8 gene. We assumed that all the genes were transcribed on 1 strand in the same direction and arranged in the same relative positions as gene loci in known cestode mitochondrial genomes. The arrangement of mitochondrial genes in Spirometra tapeworms was identical to that of other pseudophyllidean cestodes published to date, with the exception of Hymenolepis diminuta, in which the order of trnL and trnS2 is reversed. The nucleotide compositions of the whole mitochondrial genomes are 19.8% A, 45.9% T, 23.5% G, and 10.9% C (S. erinaceieuropaei) and 20.3% A, 46.0% T, 22.7% G, and 11.0% C (S. decipiens). As in other cestodes, the genomes are A + T rich: S. erinaceieuropaei, 65.7% A + T; and S. decipiens, 66.3% A + T (Table 1). Some genes overlap at the boundaries: cox3/trnH (10 bp), nad4L/nad4 (40 bp), trnQ/trnF (4 bp), trnF/trnM (4 bp), nad3/trnS1 (11 bp), and cox1/trnT (10 bp), respectively (Table 2).
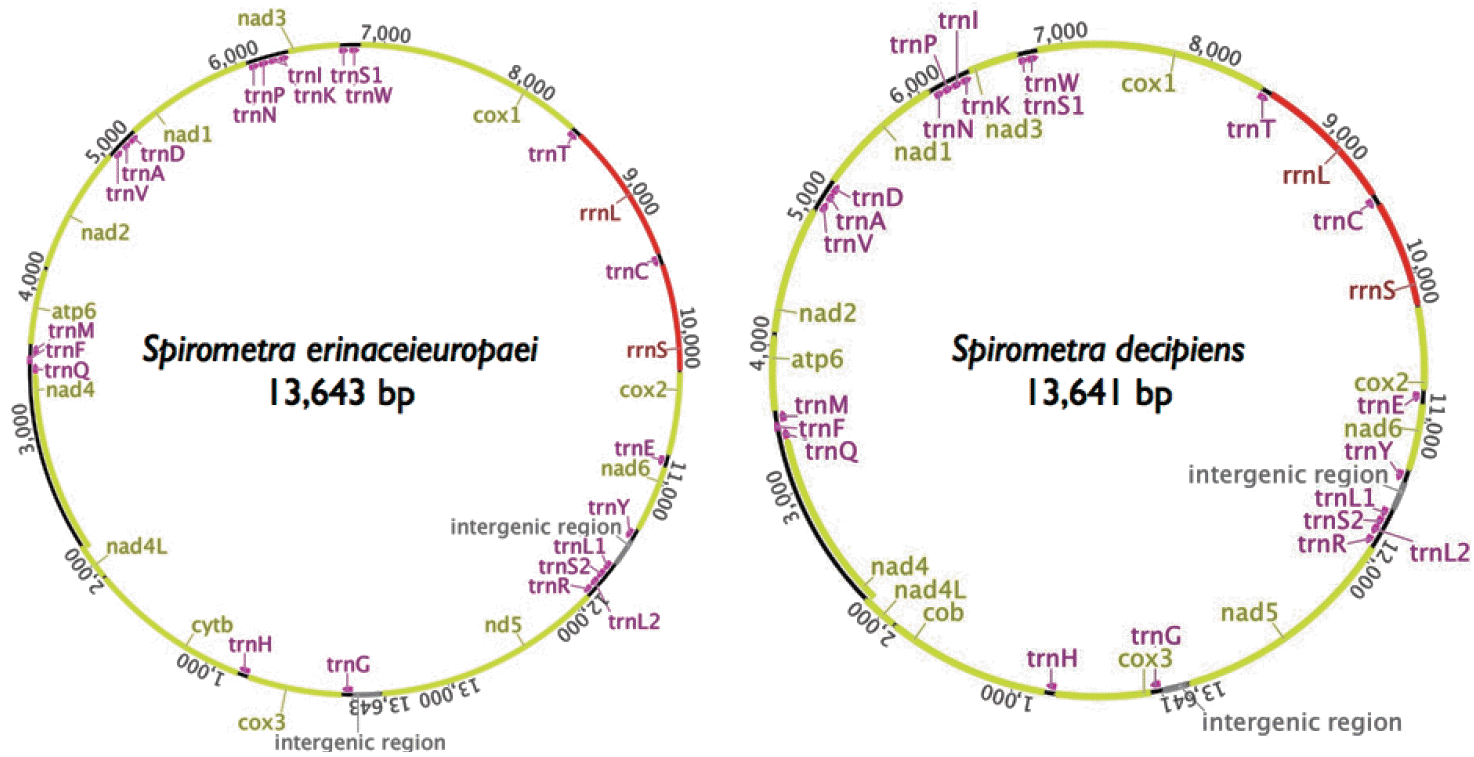
Schematic representation of the mitochondrial genomes of S. erinaceieuropaei and S. decipiens.
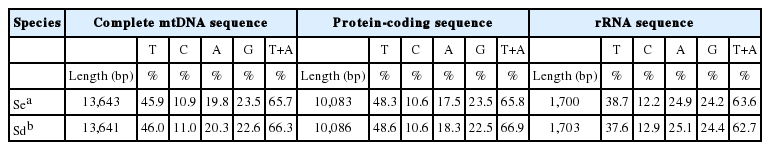
Nucleotide compositions of the complete mitochondrial genomes, protein-coding genes, and ribosomal RNA sequences of 2 Spirometra species

Position and characteristics of the protein-coding and non-coding sequences in the mitochondrial genomes of Spirometra erinaceieuropaei and S. decipiens
Protein-coding genes
Approximately 65% of the mitochondrial genomes of Spirometra tapeworms consisted of protein-encoding genes, similar to the values reported for other cestodes. The 12 protein-coding genes constituted 10,083 bp (S. erinaceieuropaei) and 10,086 bp (S. decipiens) of their respective mitochondrial genomes (Table 1). All of the putative open reading frames (ORFs) of 12 protein-coding genes in both species start and end with complete codons. The ATG initiation codon was used in 11 genes (atp6, cob, cox1, cox2, nad1, nad2, nad3, nad4, nad4L, and nad6), whereas the GTG initiation codon was used only in the cox3 gene. The TAG stop codon was used in 7 genes (cox1, cox2, cox3, nad2, nad3, nad4, and nad4L) in S. erinaceieuropaei and 6 genes (cox1, cox3, nad2, nad3, nad4, and nad4L) in S. decipiens, whereas the TAA termination codon was used in the remaining 5 genes (atp6, cob, nad1, nad5, and nad6) in S. erinaceieuropaei and 6 genes (atp6, cob, cox2, nad1, nad5, and nad6) in S. decipiens (Table 2). Codon usage is shown in Table 3. The 4 most commonly used codons were Leu (TTR and CTN; 15.6%), Phe (TTY; 12.6%), Val (GTN; 10.8%) and Ser (AGN and TCN; 9.9%) in S. erinaceieuropaei, and Leu (TTR and CTN; 15.7%), Phe (TTY; 12.5%), Val (GTN; 10.7%), and Ser (AGN and TCN; 9.9%) in S. decipiens.

Codon usage in the 12 protein-coding genes of the mitochondrial genomes of Spirometra species
Transfer RNA and ribosomal RNA genes
We identified a total of 22 tRNA genes in the S. erinaceieuropaei and S. decipiens mitochondrial genomes. The tRNA genes, ranging in length from 56 bp to 70 bp, were identified based on putative secondary structures such as the typical cloverleaf shape (Fig. 2). The inferred secondary structure of 19 tRNA exhibited the typical cloverleaf shape, with paired DHU arms; in the remaining 3 tRNAs (trnR, trnS1, and trnS2), this arm was replaced with a 7-12 nt unpaired loop. The aminoacyl acceptor arms consisted of 7 bp, and trnA, trnI, trnM, trnQ, trnR, trnS2, trnT, and trnV contained 1 or 3 non-canonical base pairs. The anticodon stems of all 22 tRNAs contained of 5 bp, as in typical stem structures. The predicted secondary structures of the 22 tRNAs in S. erinaceieuropaei and S. decipiens had paired TΨC arms, consisting of a 2-5 bp stem with a 3-9 bp loop. The variable loop between the anticodon and the TΨC stems consisted of 3-5 nt, except in trnR, trnC, trnS1, and trnS2 (Fig. 2). In both species, the ribosomal RNA genes rrnL and rrnS were separated by trnC. The rrnL and rrnS genes were 967 bp and 733 bp long, respectively, in S. erinaceieuropaei, and 973 bp and 730 bp long in S. decipiens. The nucleotide content of the rrn genes was 63.6% (A+T) in S. erinaceieuropaei and 62.7% (A+T) in S. decipiens (Table 1).
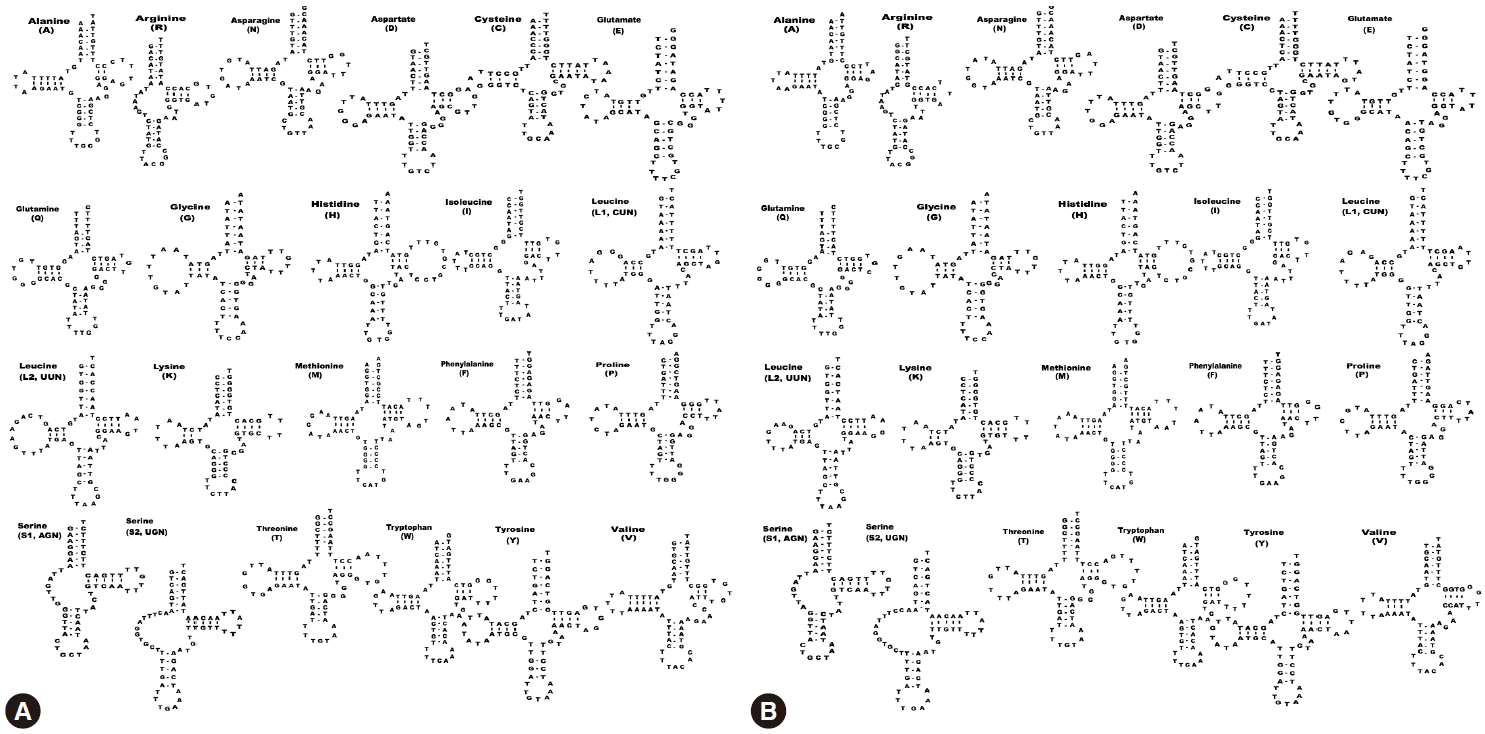
Inferred secondary structures of the 22 mitochondrial tRNA from S. erinaceieuropaei (A) and S. decipiens (B).
Non-coding regions
A total of 23 intergenic sequences, varying from 1 to 204 bp in size, were interspersed in the S. erinaceieuropaei (total, 504 bp) and S. decipiens (total, 496 bp) mtDNA. Two major non-coding regions present in the mtDNA were predicted to form hairpin structures. Non-coding region 1 (NR1), between trnY and trnL1, is 201 bp (S. erinaceieuropaei) or 204 bp (S. decipiens) in length, and non-coding region 2, between nad5 and trnG was 185 bp (S. erinaceieuroapei) or 174 bp (S. decipiens) in length. The non-coding regions in these 2 species had A + T content of 70.9% (S. erinaceieuropaei) and 67.7% (S. decipiens) and contained stem-loop structures. The nucleotide contents of the NR1 and NR2 were 32.6% A, 9.3% C, 38.3% T, and 19.7% G in S. erinaceieuropaei mtDNA, while those of S. decipiens were 32.3% A, 11.9% C, 35.4% T, and 20.4% G in S. decipiens mtDNA.
Mitochondrial sequence divergence between S. erinaceieuropaei and S. decipiens
A pairwise comparison of sequence divergence of the 12 protein-coding genes and 2 ribosomal RNA genes of S. erinaceieuropaei and S. decipiens is shown in Table 4. The protein-coding sequences of S. erinaceieuroapei contained 10,083 bp and 3,361 codons, whereas those of S. decipiens contained 10,086 bp and 3,362 codons. The 12 protein-coding genes of S. erinaceieuropaei and S. decipiens differed by 12.4%, whereas the full mtDNA sequences differed by 12.9%. Divergences in protein-coding genes between S. erinaceieuropaei and S. decipiens ranged from a low of 9.3% to a high of 13.7% (Table 4). Amino acid sequence divergences of cox1 (the most highly conserved gene) and nad6 (the most variable gene) were 1.9% and 14.8%, respectively. The ribosomal RNA genes of S. erinaceieuropaei differed by 11.2% (rrnL) and 6.9% (rrnS) relative to those of S. decipiens.
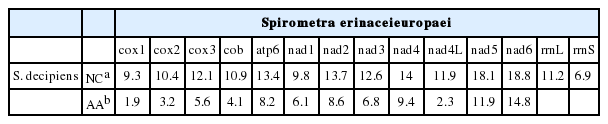
Divergences of nucleotides and amino acids of the protein-coding genes
Phylogenetic relationships of pseudophyllidean cestode among the eucestodes
We performed phylogenetic analyses of S. decipiens and S. erinaceieuropaei using 4 methods (Bayesian inference, ML, NJ, and MP), based on concatenated amino acid sequences of 12 protein genes from 11 cestodes and 2 trematodes. To this end, we used an alignment set of 10,394 bp including all 12 mitochondrial protein-coding gene loci. Of the 3,141 (30.3%) homologous positions and 66.1% pairwise identity showed in the set of those mtDNA sequences from maximum likelihood analysis. A concatenated alignment set of 3,389 homologous amino acid positions and 1,425 variable sites were phylogenetically informative under MP criterion. Phylogenetic relationships among the eucestodes determined using the 4 analytic approaches exhibited identical tree topologies. In the consensus tree, order Cyclophyllidea, including family Taeniidae (Taenia and Echinococcus) formed a well-supported monophyletic group. Family Hymenolepididae is a sister taxon to the Taeniidae. Within the Pseudophyllidea clade, Diphyllobothrium and Spirometra formed a monophyletic group, and sister genera are well supported (Fig. 3).
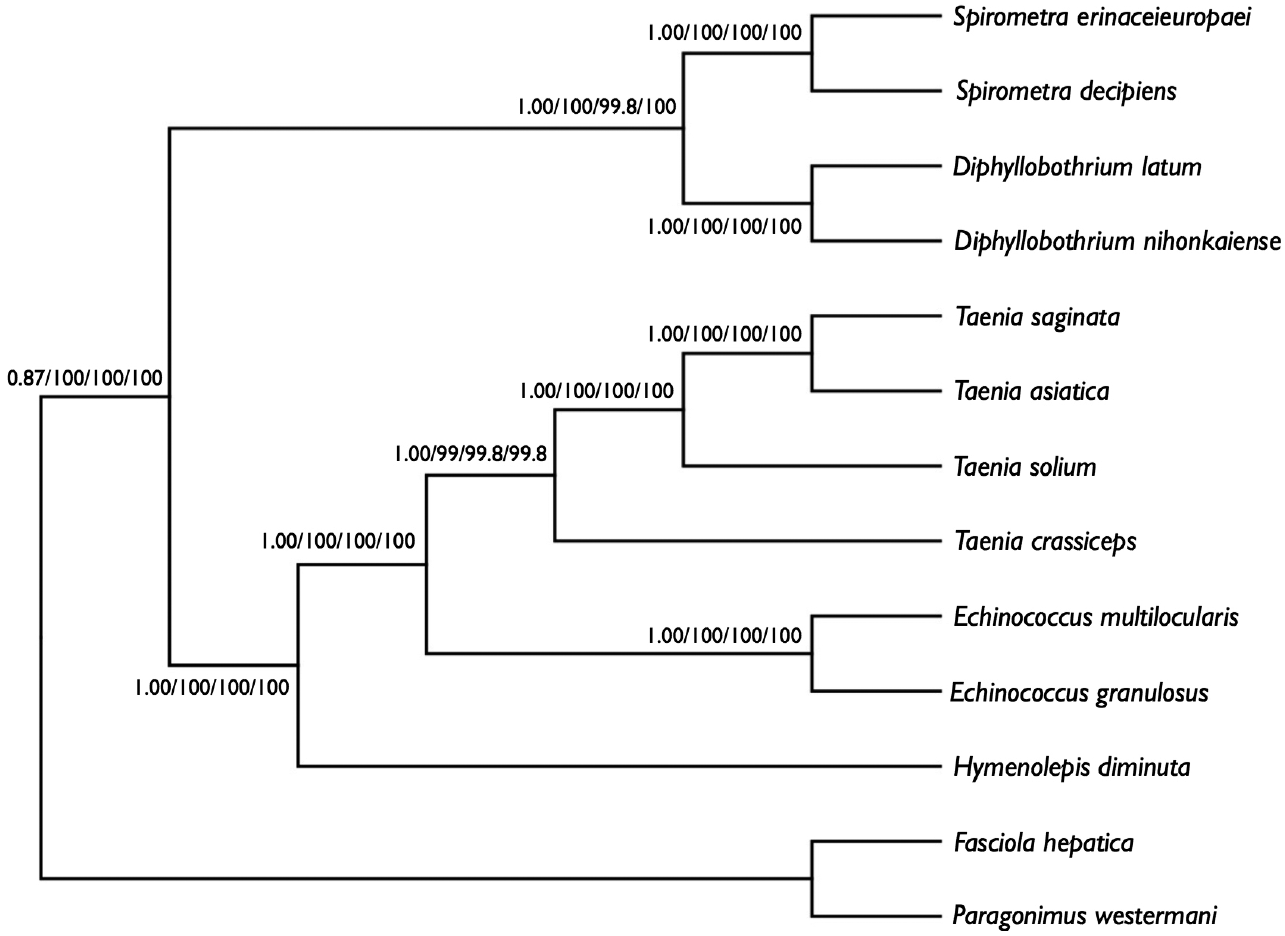
Phylogenetic relationship among eucestode species based on inferred amino acid sequence data selected from 12 mitochondrial protein-coding gene loci for 13 platyhelminthes. The numbers above the branches represent bootstrap values for Bayesian inference, maximum likelihood (ML), neighbor joining (NJ), and maximum parsimony (MP), respectively.
DISCUSSION
In the present study, we sequenced and analyzed whole mitochondrial genomes of S. erinaceieuropaei and S. decipiens based on the morphological analysis described in detail recently by the present authors [16]. The full mtDNA sequences of S. erinaceieuropaei and S. decipiens differ by 12.9% which means S. erinaceieuropaei and S. decipiens are valid species that can be distinguished from each other by comparison of mitochondrial DNA sequences and morphological data as well.
Morphologically, S. erinaceieuropaei can be clearly distinguished from S. decipiens by its spirally coiled uterus [2]. The uterus of S. erinaceieuropaei consists of 5-7 complete turns, whereas that of S. decipiens consists of 4−4½ coils. The lateral margins of the subterminal uterine coil of S. erinaceieuropaei are parallel, while those of S. decipiens are the broadest as subspherical in contour. In S. erinaceieuropaei, the uterine pore lies in the midline, a small distance behind the anterior margin of the uterine terminal ball, whereas in S. decipiens, it is a conspicuous sphincter in a ventral position under the bulge of the terminal uterine coil. The vaginal pore is a broad crescent slit behind the male genital pore in S. erinaceieuropaei, whereas in S. decipiens, it is crescent-shaped and elliptical. The cirrus is strongly muscular and elongated in shape in S. decipiens, whereas in S. erinaceieuropaei, it is smaller than in other related species [2].
The molecular characteristics of the mitochondrial genome of S. erinaceieuropaei and S. decipiens that we identified in this study, gene arrangement, nucleotide composition, genetic code, and secondary structure of tRNA, were similar to those of other cestodes. The genetic distance between S. erinaceieuropaei and S. decipiens was determined by a percentage pairwise comparison of the nucleotide and amino acid compositions of the mitochondrial genomes. The 12 protein-coding genes of S. erinaceieuropaei and S. decipiens differed by 12.4%, whereas the sequence differences for the whole mitochondrial sequences were 12.9%. Divergences of amino acid sequences between S. erinaceieuopaei and S. decipiens ranged from a low of 1.9% (cox1) to a high of 14.8% (nad6). Therefore, these 2 parasitic organisms represent distinct species within a same genus.
These species have not been clearly identified in terms of which specifically infect humans. Conventionally, S. erinaceieuropaei larval stages were considered to be the main reason for human infections through eating raw or undercooked snakes but actually the spargana were not seen at all naturally in the snakes. Rather, S. decipiens has been found from snakes (unpublished data). Natural infections with S. decipiens has been mostly observed in carnivorous animals such as cats and dogs, but no such natural infections have been observed for S. erinaceieuropaei. Thus, S. decipiens may only be found in dogs and cats (unpublished data). In this and the previous study [16], adult S. erinaceieuropaei worms were obtained by feeding dogs the larvae collected from humans or the muscle fascia of the hedgehog (Erinaceus dealbatsu); after the initial infection, adults can be harvested in about 4 weeks. The only known second intermediate host of S. erinaceieuropaei is the Chinese hedgehog. Therefore, in the context of human sparganosis, the infection route and relevant intermediate host of S. erinaceieuropaei remain unclear, and there is an epidemiological discrepancy between eating habits and distributions of Spirometra species in animals.
The information derived from the complete sequence of the S. erinaceieuropaei and S. decipiens mitochondrial genome will add to the available mitochondrial sequence data of parasitic cestodes, and provide a resource for comparative mitochondrial genome analyses of pseudophyllidean tapeworms. Our results show that S. decipiens is a valid species that can be distinguished from S. erinaceieuropaei by comparison of mitochondrial DNA sequence as well as morphological data in the previous study [16].
Acknowledgements
The corresponding author (Hyeong-Kyu Jeon) was supported by a Research Fellow Grant for young scientist from National Research Foundation of Korea (NRF-2012R1A1A2042993). This work was supported by a research grant from Chungbuk National University in 2014. Parasite materials used in this study were provided by the Parasite Resource Bank of Korea National Research Resource Center (2010-0003456), Republic of Korea.
Notes
We have no conflict of interest related to this work.