Seroprevalence of Sarcocystis falcatula in Two Islands of Malaysia using Recombinant Surface Antigen 4
Article information
Abstract
Sarcocystosis was diagnosed worldwide by serodiagnostic tests utilising the whole parasite, for which the protozoa were maintained in vitro are more costly. In this study, antigenicity of Sarcocystis falcatula recombinant protein (rSfSAG4) was investigated towards the local communities of Pangkor and Tioman Islands and its seroprevalence was surveyed in these islands. A total of 348 human sera were tested using rSfSAG4 by Western blot and ELISA. High prevalence of sarcocystosis was observed in Tioman Island (80.6%) than in Pangkor Island (50.0%) by Western blot. In ELISA, the seroprevalence observed in Tioman Island was 45.9%, whereas in Pangkor Island 63.0%. In other parasitic infections, the prevalence was 34.0% by Western blot and 46.0% by ELISA. In healthy control group, 7% by Western blot and 8% by ELISA showed positivity to rSfSAG4. It is suggested SfSAG4 is a candidate antigen to measure seroprevalence of sarcocystosis.
INTRODUCTION
Sarcocystis species are intracellular coccidian protozoan parasites causing a zoonotic disease sarcocystosis in the hosts. It has an obligatory 2-host life cycle that involves the prey-predator relationship. Infection of muscular sarcocystosis begins when the intermediate host/prey including human consumes food or water contaminated with sporocysts/oocysts. Sarcocystis invades muscle tissue and matures into sarcocysts [1].
Human sarcocystosis is considered a zoonotic infection [2]. Outbreaks of muscular sarcocystosis from 2011 to 2014 were recorded among international tourists visiting Pangkor and Tioman Islands in Malaysia. S. nesbitti utilises a snake-primate life cycle [3–5] in which human is considered an accidental host presenting intermediate host-like symptoms [6,7]. S. nesbitti DNA was detected in water samples from Tioman Island providing the evidence of the mode of transmission was via water [8].
Sarcocystosis has been reported worldwide but mostly from tropical and sub-tropical countries such as in Asia, America and Europe. In Malaysia, a seropositivity survey in 1978 was found approximately 20% of 243 persons in West Malaysia had antibodies to Sarcocystis. But, muscular or intestinal sarcocystosis infection cannot be differentiated through this seropositivity test [9]. A previous study showed that 21% out of 100 samples human tongue muscle tissues contained sarcocysts [2]. In 1993, an outbreak of sarcocystosis was reported involving American military personnel in West Malaysia [10].
There is no reported case of sarcocystosis involving local communities in the islands. Diagnosis based on clinical manifestations, stool examination and invasive procedure such as muscle biopsy are done when symptoms are being present. Most cases may go asymptomatic and rarely been reported in human. Previous studies reported serodiagnoses utilising the whole parasite, for which the in vitro cultivation is more costly and requires constant maintenance [11–13]. In this study, aim is to use the recombinant protein as the target protein to develop serodiagnosis for human sarcocystosis. Recombinant protein technology helps in rapid production of high quantity and purity of the protein and low in cost. Surface protein is antigenic and useful to develop diagnostic test [14–16]. Previous evaluation was done on S. falcatula surface antigen 3 (SfSAG3) showed the potential antigenic property that can be used to determine seroprevelance of sarcocystosis [17]. Therefore, Western blot and ELISA analyses using the (SfSAG4) may provide additional information on seroprevalence of sarcocystosis in both islands.
MATERIALS AND METHODS
Ethical clearance
The serum collection was approved by the Medical Ethics Committee of the UMMC (MECID.NO: 2015-958 and MEC ref. no: 920.16) from University of Malaya, Malaysia. The ethics for animal experiment was approved by Institutional Animal Care and Use Committee (IACUC) of the University of Malaya, Faculty of Medicine (2016-20170405/PARA/R)
PCR of SfSAG4
N-terminal signal peptide sequence and the C-terminal GPI-anchor were predicted and removed during amplification of SfSAG4. The PCR was done in 25 μl reaction volume using Promega GoTaq Flexi DNA Polymerase with 4 μl of cDNA of S. falcatula Stiles (ATCC 50701) as the template with a primer pair SfSAG4_EF (5′-AAT ACC ATA CCT CGG CGT CA-3′) and SfSAG4_ER (5′-TCA AAT GGC TGT CTC CAC AA-3′). The PCR steps were initial denaturation at 94°C for 2 min, 30 cycles of denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec, and elongation at 72°C for 90 sec. Next, final elongation at 72°C for 7 min and held at 16°C. Nested PCR was done using 2 nested primers SfSAG4_IF (5′-AAG GAT CCG GAC TTA ACC CA-3′) and SfSAG4_IR (5′-AAG GAT CCT TAA CCA CCG GCC G-3′), where BamHI restriction site in bold. The primary PCR product, 4 μl was added to the nested PCR mix and thermal cycle was the same except annealing at 58°C for 30 sec.
Cloning and production of recombinant SfSAG4 protein
The nested PCR amplicon SfSAG4 was ligated into pRSET A vector and transformed into E. coli BL21(DE3)pLysS. A recombinant plasmid was sequenced and analysed by BLAST. The SfSAG4 expression was induced by adding 1 mM IPTG at 37°C and shaking at 250 rpm for 3 hr. The recombinant SfSAG4 fusion protein was purified using BugBuster Master Mix and 8 M urea under denaturation. The fusion protein was refolded by dialysis against gradient urea-PBS, and then subjected to 12% SDS-PAGE and Western blot analyses.
Production of immune sera
The BALB/c mice, female, 2–4-week old, were divided into 2 groups: immunised group (rSfSAG4 and SfTLA) and control group (protein of pRSET A and BTTLA) (n=2 per group). For the purified proteins, 30–100 μg of protein was mixed with adjuvant in volume of 1:1 ratio and the mixture was injected into each mouse in a final volume of 100 μl. Complete Freund’s Adjuvant (Sigma Chemical Co., St Louis, Missouri, USA) was used in the primary boost and Incomplete Freund’s Adjuvant Sigma Chemical Co.) was used in the subsequent boosters. Booster was given on day 14 and 21 post-immunisation. All injections were given subcutaneously. All mice were sacrificed 10 days after second booster (day 31 post-immunisation) and the intracardiac puncture was performed to withdraw blood. Blood was clotted and centrifuged at 3,000 rpm for 20 min. Serum was collected and stored at −20°C until use.
Serum collection
A total of 198 sera were collected from villagers in the two islands and 150 sera of parasitic infections patients and healthy donors were collected from University of Malaya Medical Centre (UMMC).
Detection of anti-rSfSAG4 antibody in human sera using Western blot
The rSfSAG4 protein (70–100 ng) was blotted onto PVDF membrane for each serum sample. The membrane was blocked in 2.5% skimmed milk overnight at 4°C. The incubation steps are as follow: serum sample (1:250 dilution in 2.5% skimmed milk for 2 hr at room temperature), biotin-labelled goat anti-human IgM+IgG+IgA (1:2,500 dilution in 2.5% skimmed milk/TBS for 1 hr), streptavidin-alkaline phosphatase (1:2,500 dilution in 2.5% skimmed milk/TBS for 1 hr) and NBT/BCIP substrate.
Detection of anti-rSfSAG4 antibody in human sera using ELISA
The rSfSAG4 protein, 10 μg/ml, in 100 μl of 0.05 M sodium bicarbonate buffer (pH 9.6) was coated on 96-well microtitre plate (Nunc MaxiSorp, Thermo Scientific, Massachusetts, USA) and incubated overnight at 4°C. The incubation steps were as follow: 1% BSA/PBS blocking for 2 hr at 37°C, and serum sample (1:80 dilution in 1% BSA/PBS) for 1 hr at 37°C, HRP-labelled goat anti-human IgM+IgG+IgA (1:2,500 dilution in 1% BSA/PBS) for 1 hr at 37°C. 3, 3′,5, 5′-Tetramethylbenzidine (TMB) was added to the reaction and kept for 30 min in dark and stopped with 2 N sulphuric acid. Absorbance of each sample was read at 450 nm. A cut-off value was set with mean absorbance +2 SD of the control serum samples.
RESULTS
Identity of SfSAG4
BLAST result of the nucleotide and amino acid sequences of rSfSAG4 were 82% and 78% identity to reference sequence of S. falcatula (GQ851959.1 and ADG26782.1), respectively. SfSAGs are polymorphic; hence the lower identity coverage with the reference sequences was expected [18,19].
Purified rSfSAG4
The purified rSfSAG4 was detected with an expected molecular weight at −27 kDa (Fig. 1).

Detection of purified rSfSAG4 proteins in Coomassie blue-stained SDS gel.
Antigenicity of rSfSAG4
Antigenicity of rSfSAG4 was evaluated against mouse anti-rSfSAG4 and anti-SfTLA immune sera. A single band of rSfSAG4 (−27 kDa) was observed when probed with mouse anti-rSfSAG4 and anti-SfTLA immune sera (Fig. S1).
Seroprevalence of anti-rSfSAG4 antibody
Serprevalence of sarcocystosis falcatula was 80.6% in Tioman Island and 50.0% in Pangkor Island by Western blot. In ELISA, the prevalence rate observed 45.9% in Tioman Island, and 63.0% in Pangkor Island. In other parasitic infections, the antibody positive rate was 34.0% by Western blot and 46% by ELISA. In the healthy control group, antibody positive rate was 7% by Western blot and 8% by ELISA (Fig. 2).
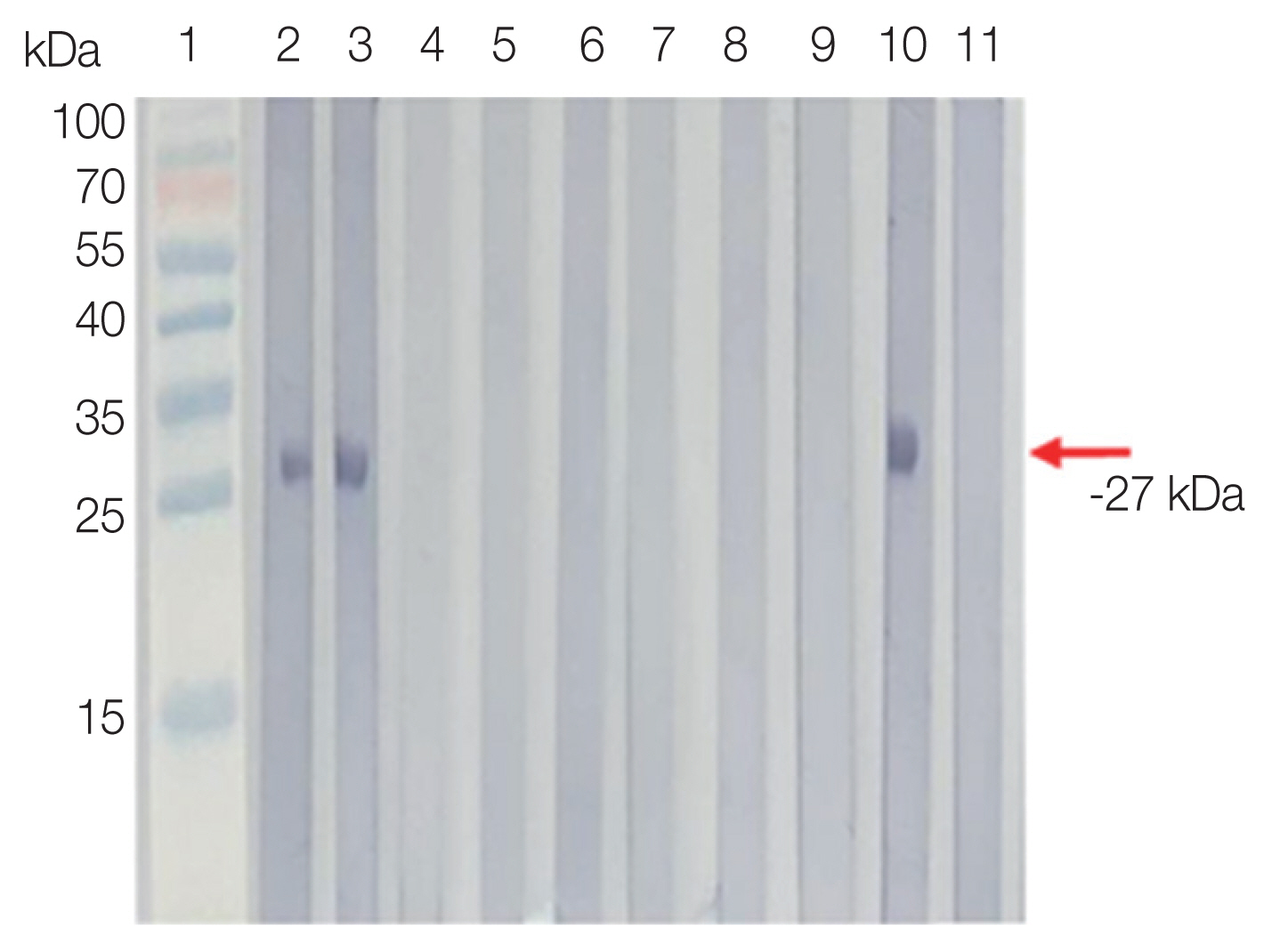
Western blot analysis of rSfSAG4 probed with selected serum of volunteers from Pangkor and Tioman Islands (lanes 2 and 3), patients infected with non-Sarcocytis parasitic infections (lanes 4 to 9), positive control (lane 10) and healthy donors (lane 11).
To cross-check the positive reactivity, the selected positive sera of volunteers and healthy donors were tested with S. falcatula TLA. A single band of −24 kDa was observed for positive samples and no band for the negative control (healthy donor) (Fig. S2).
DISCUSSION
As mentioned earlier, the aim of this study is to evaluate rSfSAG4 as protein candidate for serodiagnosis of sarcocystosis. This is a continuation study from our previous work on rSfSAG3, where rSfSAG3 showed immunodominant characteristic and a suitable protein candidate for the development of serodiagnostic tools. The experimental design and the type of tested sera were the same as our previous work [17]. We were unable to test sensitivity and specificity of rSfSAG4 due to absence of true positive serum (patient infected with S. nesbitti) in this study. In addition, previously obtained S. nesbitti DNA failed to amplify when tested with SAG4 gene-specific primers, hence the use of S. falcatula DNA as the replacement to amplify the gene [8]. For this reason, we can only use SfSAG4 gene isolated from S. falcatula. The nucleotide sequence was available from GenBank during the course of study.
Positive sera showed antibody response to rSfSAG4 and SfTLA suggesting antigenic immunodominancy of the rSfSAG4.
Similar finding in rSfSAG3, sera from Tioman Island showed higher reactivity towards rSfSAG4 then in Pangkor Island in Western blot. Higher risk factor such as the main use of untreated water may contribute to the difference in seroprevalence [20]. Additionally, previous Sarcocystis infection is also a possible reason. The discrepancy of Western blot and ELISA was expected, where absorbance value of many sera in ELISA were in borderline of positives or negatives. Similar finding was reported in previous study on rSfSAG3. On the other hand, 13% of the T. gondii infected patients and 56.5% of the malaria infected patients were reacted positively in Western blot. In ELISA, 41% of the T. gondii infected patients and 60.8% of the malaria infected patients were reacted positively. Co-infection of Sarcocystis and Toxoplama was reported in Malaysia although no report on the co-infection between Sarcocystis and Plasmodium [9,21]. Similar to rSfSAG3 evaluation [17], rSfSAG4 was tested with anti-T. gondii and anti-Plasmodium via Western blot to eliminate potential cross-reactivity. Additionally, sequence of rSfSAG4 was subjected to BLAST analysis to check for the potential similarity with the antigens for other parasites resulted in no significant similarity. Similar risk factors play a major role in co-infection although the study of Sarcocystis with other parasites is inadequate.
SfSAG4 is a candidate antigenic protein to measure seroprevalence of sarcocystosis. With further evaluations and validation of the serodiagnosis tests with true positive sera, SfSAG4 can potentially be combined with SfSAG3 as a chimeric SfSAG3/4 fusion protein for a more sensitive diagnostic agent since the previous work on rSfSAG3 showed immunodominant characteristic of SfSAG3.
Supplementary Information
rSfSAG4 probed with mouse anti-rSfSAG4 (A), anti-SfTLA (B, lane 1) and anti-BTTLA (B, lane 3) immune sera.
Reactivity of (A) S. falcatula total lysate with a selected positive serum and healthy donors by Western blot.
ACKNOWLEDGMENTS
We would like to thank Prof. Dr. Rohela Mahmud from Department of Parasitology, Faculty of Medicine, University of Malaya for the kind support for sample collection. This study was supported by the University of Malaya High Impact Research Grant (UM.C/HIR/MOHE/MED/16) from the Ministry of Higher Education, Malaysia, Postgraduate Research Fund (PG033-2016A) from University of Malaya and Bantuan Kecil Penyelidikan fund (BKS001-2017) from University of Malaya.
Notes
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this study.