Abstract
Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1), encoded by the polymorphic var multigene family, is a highly polymorphic antigen that plays a crucial role in the pathology of malaria. The contribution of the genetic diversity of var toward the immune escape of P. falciparum has not yet been fully elucidated. This study aimed to characterize the diversity of var repertoires by screening P. falciparum Duffy-binding-like α domain (PfDBLα) among field isolates from central Myanmar. Genetic analysis revealed that the D-H segments of var in Myanmar populations have an extensive polymorphic repertoire, with high numbers of unique sequence types in each individual. However, var genes from the global population, including Myanmar, shared close genetic lineages regardless of their geographic origins, indicating that they have not undergone rapid evolutionary changes.
-
Key words: Plasmodium falciparum, erythrocyte membrane protein 1, genetic diversity, Myanmar
Introduction
Malaria, one of the most serious vector-borne infections, is caused by
Plasmodium parasites. In 2020, 241 million people were diagnosed with malaria, of which 627,000 died. Half of the world’s population is at risk of contracting the disease [
1]. Malaria parasites exploit the diversity of their surface antigens to evade the host’s immune system, which is a major impediment to the development of effective malaria vaccines [
2].
Several
Plasmodium species contain multigene families encoding variant surface antigens involved in cell binding.
P. falciparum erythrocyte membrane protein 1 (
PfEMP1), encoded by the polymorphic var multigene family, is a well-characterized variant antigen expressed on the surface of infected erythrocytes and implicated in the pathology of malaria [
3]. Although
PfEMP1 is highly variable among parasite isolates, only one type is expressed on the surface of infected red blood cells. It is either stably inherited through successive cell cycles or switched off by the expression of a different gene during infection [
4,
5].
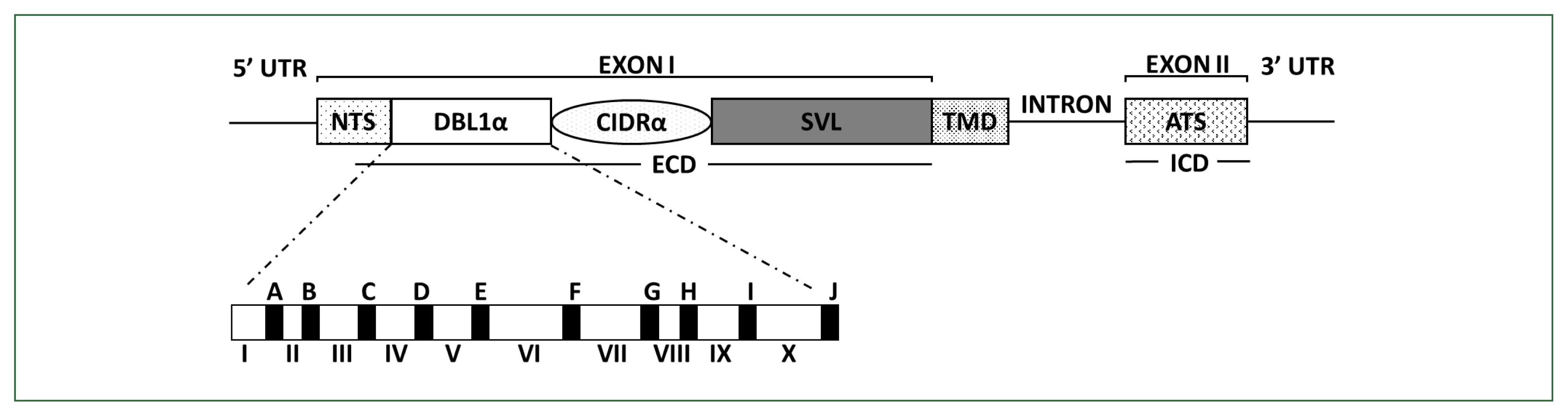
PfEMP1 comprises domain assemblies that vary in their composition and sizes and is arranged in intra- and extracellular regions. Each member of the multicopy
var family contains 2 exons (
Fig. 1). Exon 2, approximately 1.5-kb long, is highly conserved and encodes a putative intracellular region called the acidic terminal segment, whereas exon 1, approximately 4–10-kb long, is highly variable and encodes a putative extracellular region and a transmembrane domain [
6]. The extracellular region predominantly comprises an N-terminal segment, a duplicated arrangement of Duffy-binding-like (DBL) domains, tandem cysteine-rich interdomain regions (CIDRs), and C2 domains. The DBL and CIDR domains form 5 (α, β, γ, δ, and ɛ) and 3 (α, β, and γ) types, respectively, in which sequences have unique specific consensus motifs that can be used to characterize distinct DBL-CIDR domains. The prototypical head structure of most
PfEMP1 members includes a DBL1α-CIDR1α semi-conserved tandem, followed by DBLδ-CIDRβ [
6–
10].
Malaria distribution in Myanmar is heterogeneous and is influenced by several factors, such as the environment, population geospatial repartition, and international and internal migration [
11–
13]. Some molecular epidemiological studies have been conducted on
P. falciparum isolates collected from the border areas of Myanmar [
14,
15]; however, information on the genetic diversity of
PfEMP1 among isolates from other regions of Myanmar is lacking.
This study aimed to characterize the genetic diversity of var repertoires. We screened PfDBLα in P. falciparum isolates collected from central Myanmar. Understanding the nationwide diversity of PfEMP1 can provide insights into the genetic structure of the parasite and the root of persistent disease transmission, ultimately aiding in the successful elimination of the parasite.
Materials and Methods
Ethics statement
This study was approved by the Ethics Committee of the Ministry of Health, Myanmar (97/Ethics 2015) and Kyungpook National University (2017–0010). Written informed consent was obtained from each participant. For individuals aged <18 years, consent was obtained from their parents.
Study sites and sample collection
Samples were collected from the central areas of Myanmar, including Mandalay, Naung Cho, Pyin Oo Lwin, and Tha Beik Kyin, from patients diagnosed with
P. falciparum infections between August 2013 and December 2015. Falciparum malaria was diagnosed using a rapid diagnostic test and a subsequent microscopic examination followed by polymerase chain reaction (PCR) for confirmation [
16]. A total of 48 blood samples from
P. falciparum-infected patients (42 men and 6 women, age 3–50 years) were analyzed in this study (
Supplementary Table S1). Genomic DNA (gDNA) was purified from the samples using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany).
PfDBLα was amplified from the gDNA of each isolate using previously used degenerate primer sets BF (forward: 5′-GCMTGYGCDCCRTWYMGAMG-3′) and BH (reverse: 5′-CKGCCCATTCYTCRAACCA-3′) targeting the semi-conserved blocks B and H of DBLα [
3,
17]. Briefly, PCR was performed with the following conditions: 94°C for 2 min (initial denaturation); 35 cycles at 94°C for 5 sec, 50°C for 20 sec, and 60°C for 45 sec; and 60°C for 2 min (final extension) [
18]. The PCR results were confirmed by electrophoresis on a 1.5% agarose gel, followed by staining with ethidium bromide and visualization under ultraviolet light.
Cloning and sequencing
For each isolate, amplicons scattered in the 450–700-bp region were gel-purified, ligated into the pGEM-T Easy Cloning Vector Kit (Promega, Madison, WI, USA), and transformed into Escherichia coli DH5α competent cells according to the manufacturer’s instructions. Twenty to forty colonies per each cloned amplicon were selected and sequenced using Sanger’s method (Macrogen Inc., Daejeon, Korea). The obtained sequences were aligned with global sequences from GenBank and analyzed using CLC Main Workbench 6.
Sequence analysis
Var sequence data from the global population
The global
var sequence data used for the comparison study were downloaded from GenBank and corresponded to 3 regions: Southeast Asia, Sub-Saharan Africa, and South America. In total, 73 sequences were obtained from Papua New Guinea, Thailand, and Indonesia (Asian population); Gabon, Kenya, Mali, Malawi, Sudan, Tanzania, and Senegal (African population); and Brazil, Venezuela, and Honduras (South American population). Additionally, 38
var sequences from the 3D7 strain were obtained from PlasmoDB. Accession numbers for the downloaded sequences are listed in
Supplementary Table S2.
The phylogenetic tree was constructed using MEGA X (
https://www.megasoftware.net/) with the minimum-evolution method and the maximum composite likelihood model. Evolutionary analyses were performed on the Myanmar
var sequences using the 3D7 strain and global populations as references.
Results
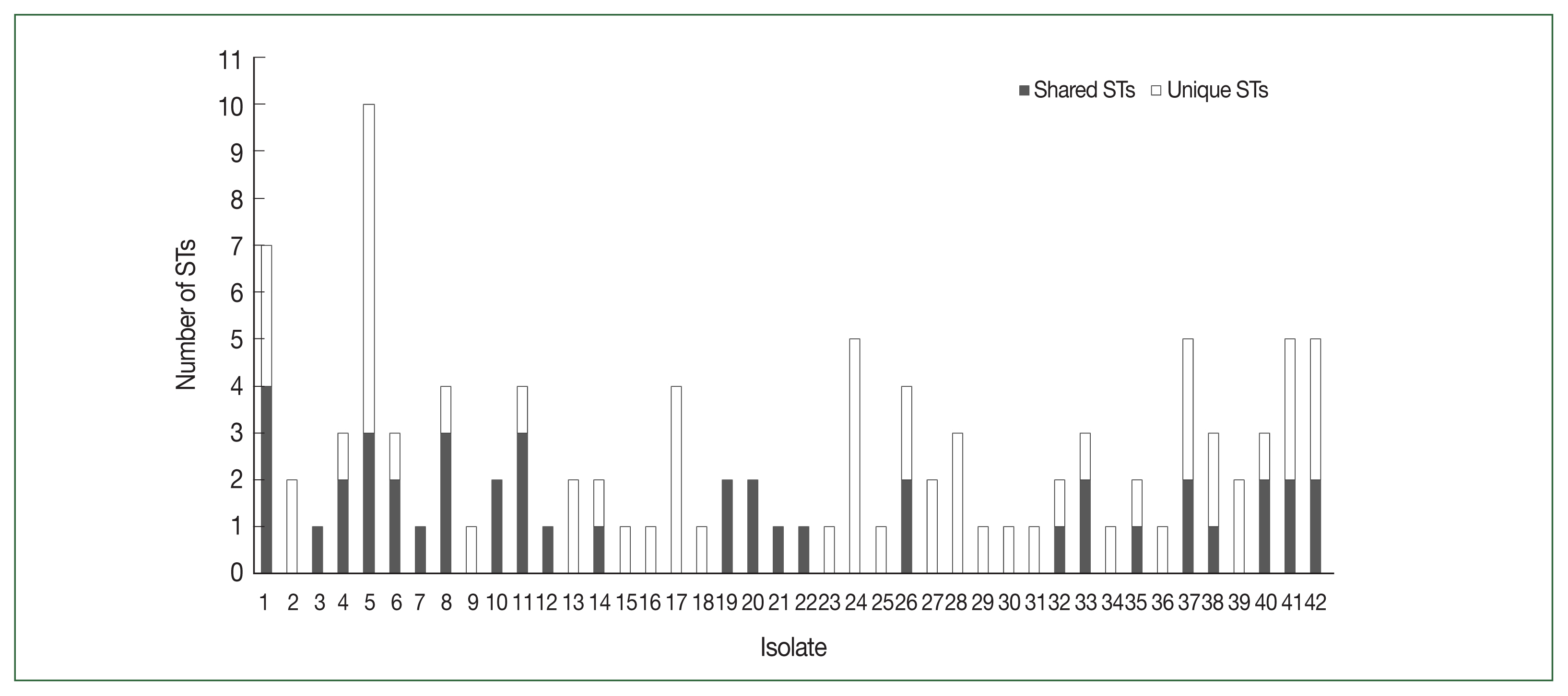
PfDBLα sequence types
Of the 48 Myanmar
P. falciparum isolates, 42 successfully yielded DBLα reads. After applying the exclusion criteria, 1033 individual BF/BH inserts were successfully sequenced, resulting in 82 distinct DBL STs (
Table 1). The
PfEMP1-encoding sequences obtained have been deposited in GenBank (Accession Nos. ON991795–ON991950). Most isolates had multiple STs with unique sequences that occurred only once in an isolate (singletons). The number of distinct DBL
var sequences detected per isolate varied from 1 to 20. The distribution of STs in the study population is shown in
Fig. 2.
The DBLα sequence from each isolate was aligned against the sequences hosted on the 3D7 genome, and the sequence with the highest alignment score was assigned the name of the gene as identified from the 3D7 genome (
Table 2). Thirty-nine different 3D7 homologs containing
PfDBLα-encoding sequences were identified. Transcripts from the DBLα groups A, B, C, B/A, and B/C were detected in isolates, and those from groups B and C were mostly shared. The highly dominant DBLα sequences were confirmed to belong to
var B, including the transcripts PFA0005W (33.3%), PFD0005W (26.2%), and PFB0010W (21.4%), followed by
var C, including the transcripts PFD0630C (23.8%) and PFD0615C (21.4%;
Table 2).
The genetic diversity of the studied population was compared by aligning against the D-H segment of the
P. falciparum 3D7 var genes. All individuals with DBLα exhibited conserved cysteine-rich motifs at all positions (
Supplementary Fig. S1).
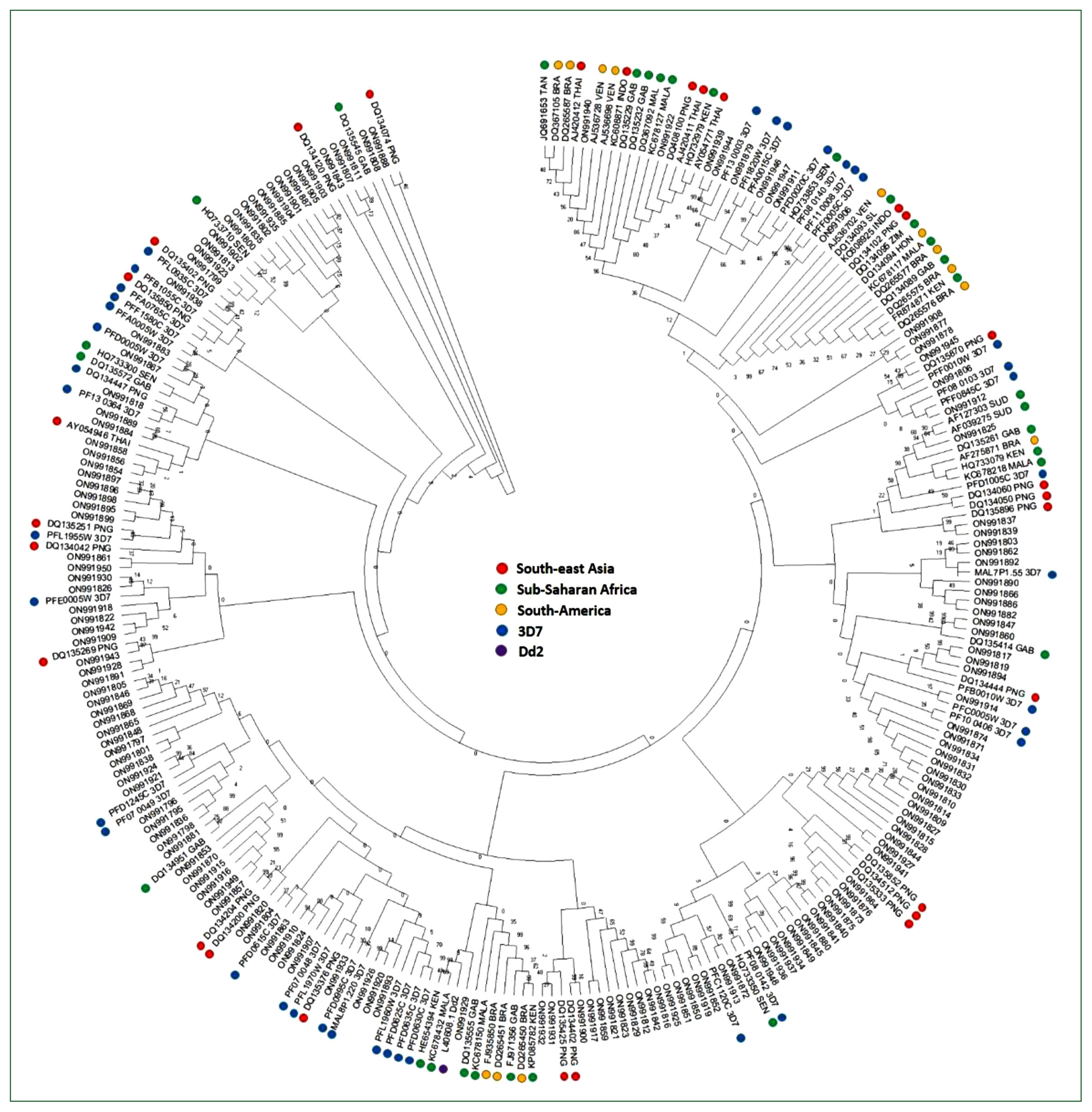
Var sequences from the Myanmar population were highly diverse and clustered with the global var genes used as a reference by sharing the bootstrap root from the Asian, European, African, and American
var sequences (
Fig. 3). In addition, the D-H segment sequences of var genes from all continents (Asia, Europe, Africa, and America) and the 3D7 strain were distributed in all branches without any region-specific cluster.
Discussion
Myanmar bears the high burden of malaria in the Greater Mekong Subregion. Although the recent incidence of malaria in the country has decreased, the genetic diversity of malaria parasites remains high, making it an important public health problem. Asymptomatic infections and the spread of antimalarial drug resistance also are major concerns [
13,
15]. In addition, the level of malaria transmission in Myanmar varies by region, making the regional persistence of the disease heterogeneous [
19–
22]. Therefore, understanding the genetic diversity of malaria parasites represents a major step toward disease eradication because it can provide insights into effective vaccine development.
In this study, we investigated the genetic diversity of var genes within a natural
P. falciparum population in central Myanmar by analyzing their DBLα domain-encoding sequences. This particular sequence motif was chosen because DBLα domains are used as polymorphic markers in gene diversity and gene expression studies. Moreover, hyperendemic regions, such as DBLα domains, directly help the parasite evade the immune response [
17,
23,
24]. A high degree of
var recombination increases the genetic diversity between isolates in neighboring and distant geographic locations [
6,
25]. Therefore, examining the genetic diversity of DBLα-encoding sequences can provide valuable information about the molecular structure of the
P. falciparum population in Myanmar. An extensive range of
var repertoires was found among the studied population. After applying a 96.0% cutoff to define the DBLα types with a minimum sequence overlap, 106 STs were identified, and singletons occurred at a high frequency in the population (
Fig. 3). The mean number of shared ST sequences among isolates was not significant, suggesting a constant event in the
var recombination repertoire and extensive diversity.
The
var-encoded hypervariable
PfEMP1 family of proteins mediates the adhesion of infected erythrocytes to various host cells during the blood stage of malaria infection. Most
PfEMP1-encoding var genes are located in the subtelomeric regions, whereas others are located centrally in chromosomes [
26]. Based on their sequence similarity and analysis of the intron and 5′ and 3′ untranslated regions, var genes can be categorized into 3 major subgroups (
var groups A, B, and C) and 2 intermediate groups (B/A and B/C), which represent transitions between the 3 main groups [
27]. Cham et al. reported that children progressively acquired a broader repertoire of anti-
PfEMP1 antibodies in the background of high exposure to malaria infection, but as their age increased, the identified antibodies acquired against particular DBL-like domains belonged to groups other than A or B/A [
28]. Therefore, in individuals frequently exposed to malaria, parasites express higher levels of groups B, C, or B/C
PfEMP1, which correlates with their reduced fitness and pathogenicity during subsequent infections [
28]. The population examined in this study was within the working age (93.0%), and
var group B predominated, followed by
var group C (
Table 2). The expression of a restricted and antigenically semi-conserved subset of
PfEMP1 suggests that this population has acquired a minimum level of immunity against malaria infection because of being constantly exposed to the disease.
In this study, within individual patients, several DBL α sequences were found more frequently than others but they were also shared between individuals. The DBLα domain contains 16–18 conserved cysteine residues, which likely play an important role in its folding and structure [
29]. The number of cysteine residues determines the classification of the DBLα domain and the severity of the disease [
6,
30]. Polymorphism within the hypervariable region induced via gene recombination and/or duplication results in length and sequence variability among parasites between and within hosts in malaria-endemic areas [
31]. The DBLα domain has an average of 4 cysteine residues, and the number can vary from 2 to 9. Variants with unusual numbers of residues may exhibit altered antigenic and adhesive properties. Among the Myanmar population studied, the cysteine at the CRC motif was almost conserved in all the var genes and was highly conserved in the homology blocks VWKA
iTC and FR
kTC. The semi-conservation of cysteine residues within the Myanmar sequences suggests that cysteine contributes toward parasite maintenance in these low-endemic areas. Even with the low number of patients included in this study, the targeted DBL1α domain showed a global distribution of
var within the central regions of Myanmar. On account of sharing borders with China, India, and Thailand, we expect to find predominant geographical
var types related to any or all of these countries. However, these results highlight the constancy of population movement in and out of different countries that share borders with Myanmar [
32]. Consistent with the conserved cysteine residues among populations, phylogenetic analysis of var genes from Myanmar and global isolates revealed that the D-H segment was not distributed in region-specific clusters, although the gene showed genetic diversity within isolates.
Notes
-
The authors declare no conflict of interest related to this study.
-
Author contributions
Conceptualization: Chung DI, Hong YC, Na BK, Goo YK
Data curation: Dinzouna-Boutamba SD, Lee S, Goo YK
Formal analysis: Dinzouna-Boutamba SD, Lee S, Moon Z
Funding acquisition: Goo YK
Investigation: Dinzouna-Boutamba SD, Lee S, Na BK
Resources: Chung DI, Myint MK, Naw H, Na BK
Validation: Naw H,
Visualization: Lee S
Writing – original draft: Dinzouna-Boutamba SD, Lee S, Goo YK
Writing – review & editing: Na BK, Goo YK
Supplementary Information
Acknowledgment
This research was financially supported by the Basic Science Research Program (NRF-2019 R1C1C1002170) and Brain Pool program (NRF-2018H1D3A1A02074759) funded by the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea (NRF).
Fig. 1Schematic overview of PfEMP1 and DBLα domain structures encoded by var. PfEMP1 comprises an extracellular domain (ECD), a transmembrane domain (TMD), and an intracellular acidic terminal segment (ATS). The ECD varies the most in its composition and size. In the DBL1α domain, conserved sequences are labeled from A to J (filled blocks), and hypervariable sequences are labeled from I to X (unfilled blocks), respectively. PfEMP1, Plasmodium falciparum erythrocyte membrane protein 1; CIDR, cysteine-rich interdomain region; DBL, Duffy-binding-like domain; NTS, N-terminal segment; SVL, segment of variable length.
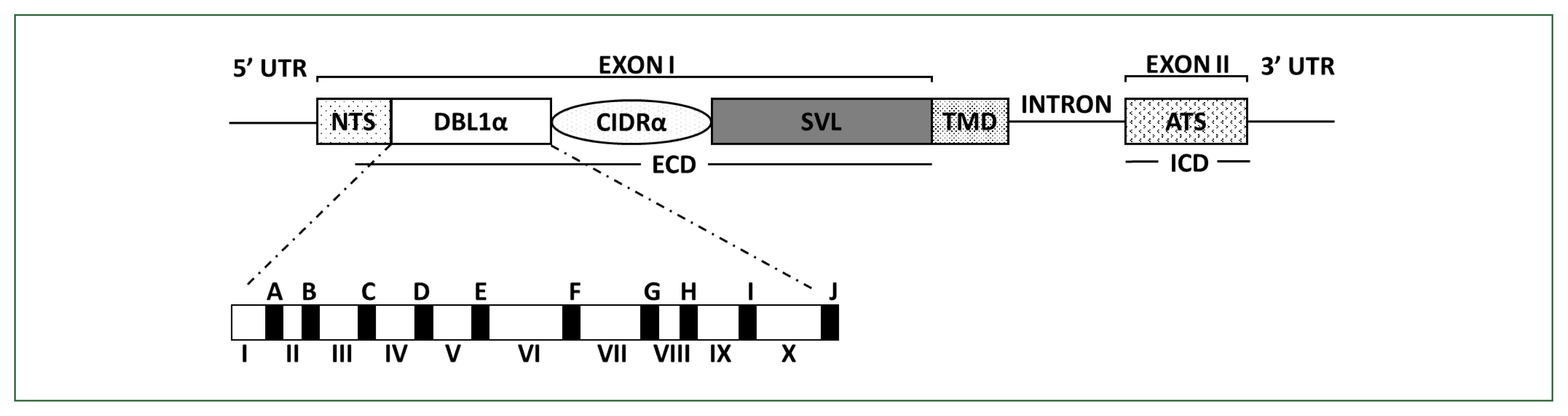
Fig. 2Distribution of PfDBLα variants in 42 Myanmar isolates. PfDBLα nucleotide sequences with at least 96.0% identity were classified into the same sequence type (ST) of var. Shared STs, sequences shared between isolates; Unique STs, sequences occurring only once within isolates.
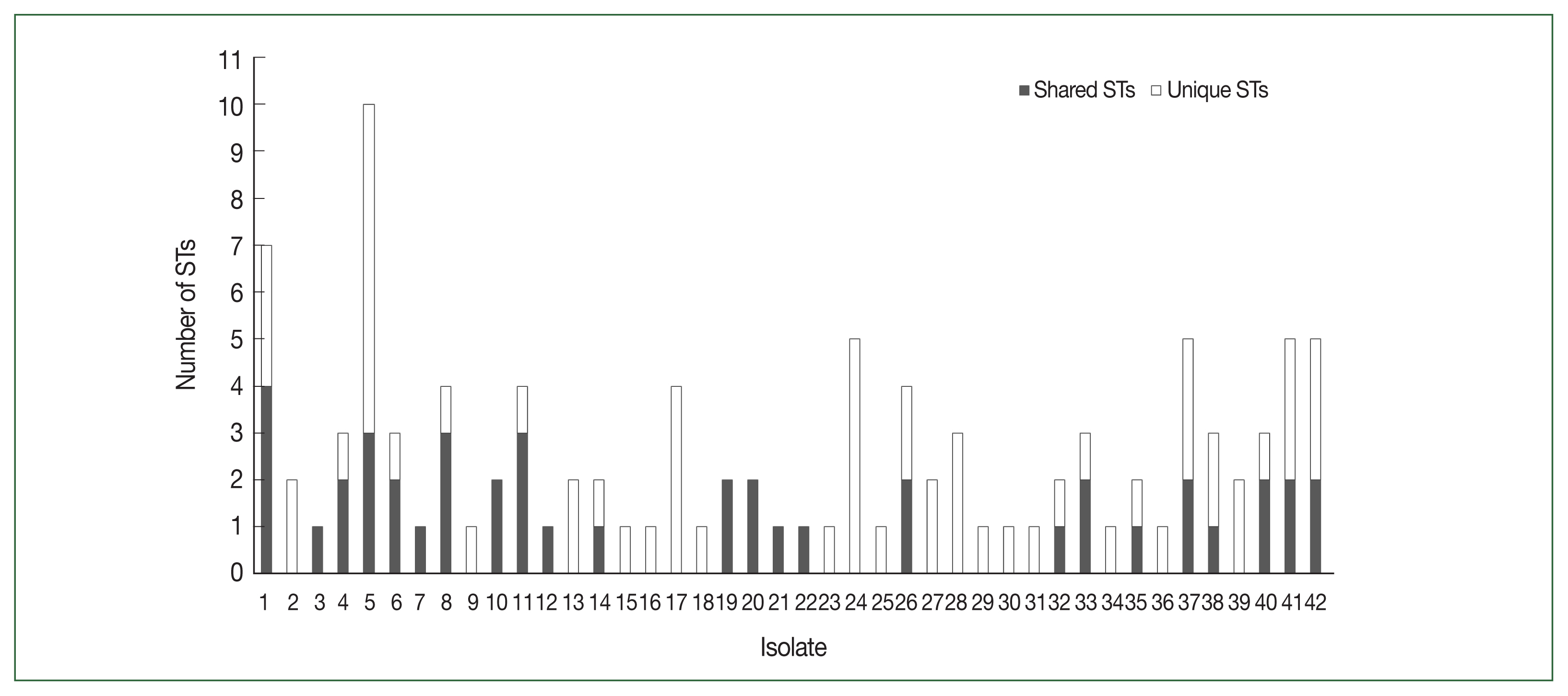
Fig. 3Phylogenetic tree of the var of Myanmar and global P. falciparum populations. A minimum-evolution tree was constructed using MEGA X with pairwise alignment and 1,000 bootstrapping repetitions.
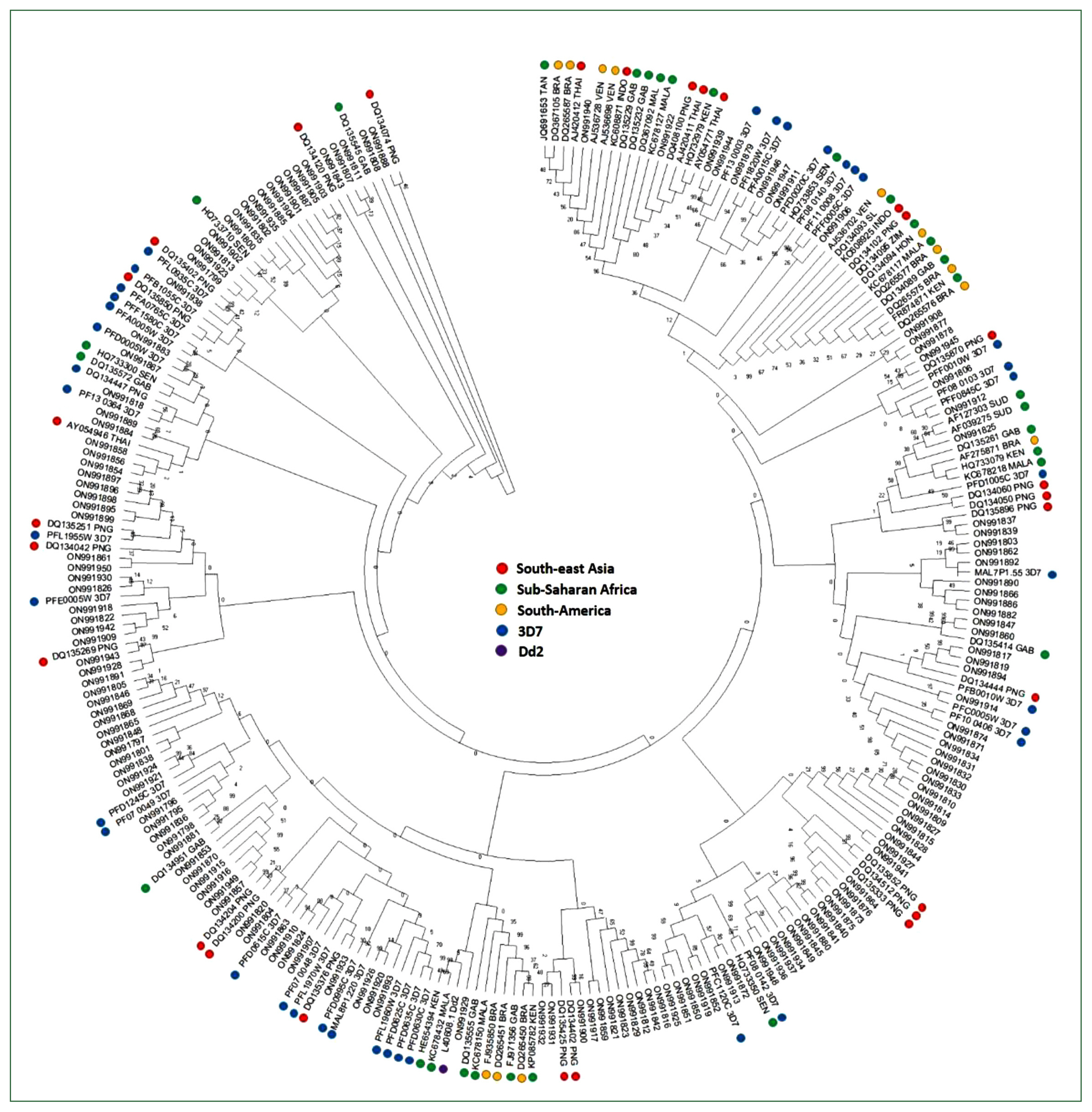
Table 1Summary of the sequencing results for Myanmar isolates
Table 1
|
Parameter |
Value |
Values after trimming and exclusion |
|
Total sequence reads from the Myanmar isolates |
1,121 |
1,033 |
|
Total no. of isolates with sequences reads |
44 |
42 |
|
No. of isolates with DBLα sequence reads |
42 |
42 |
|
Mean no. of isolates with DBLα sequence reads per isolate |
4 |
3 |
|
Median no. of isolates with DBLα sequence reads per isolate |
5 |
6 |
|
No. of DBLα sequence types |
106 |
82 |
|
Median no. of DBLα sequence types reads per isolate |
4 |
2 |
|
No. of unique DBLα types or singletons in the sample |
68 |
50 |
Table 2Summary of predominant DBLα sequences
Table 2
|
Transcript blasted (Var group) |
No. of isolates (% among studied isolates) |
No. of sequences |
|
PFA0005W (Var B) |
14 (33.3) |
20 |
|
PFD0005W (Var B) |
11 (26.2) |
20 |
|
PFD0630C (Var C) |
10 (23.8) |
12 |
|
PFB0010W (Var B) |
9 (21.4) |
18 |
|
PFD0615C (Var C) |
9 (21.4) |
15 |
|
PFB1055C (Var B) |
7 (16.7) |
9 |
References
- 1. World Health Organization. World Malaria Report 2021. World Health Organization; Geneva, Swithzerland. 2021.
- 2. Deitsch KW, Dzikowski R. Variant gene expression and antigenic variation by malaria parasites. Annu Rev Microbiol 2017;71:625-641. https://doi.org/10.1146/annurev-micro-090816-093841
- 3. Taylor HM, Kyes SA, Harris D, Kriek N, Newbold CI. A study of var gene transcription in vitro using universal var gene primers. Mol Biochem Parasitol 2000;105(1):13-23. https://doi.org/10.1016/s0166-6851(99)00159-0
- 4. Tembo D, Montgomery J. Var gene expression and human Plasmodium pathogenesis. Future Microbiol 2010;5(5):801-815. https://doi.org/10.2217/fmb.10.33
- 5. Jensen AR, Adams Y, Hviid L. Cerebral Plasmodium falciparum malaria: the role of PfEMP1 in its pathogenesis and immunity, and PfEMP1-based vaccines to prevent it. Immunol Rev 2020;293(1):230-252. https://doi.org/10.1111/imr.12807
- 6. Smith JD, Gamain B, Baruch DI, Kyes S. Decoding the language of var genes and Plasmodium falciparum sequestration. Trends Parasitol 2001;17(11):538-545. https://doi.org/10.1016/s1471-4922(01)02079-7
- 7. Peterson DS, Miller LH, Wellems TE. Isolation of multiple sequences from the Plasmodium falciparum genome that encode conserved domains homologous to those in erythrocyte-binding proteins. Proc Natl Acad Sci U S A 1995;92(15):7100-7104. https://doi.org/10.1073/pnas.92.15.7100
- 8. Baruch DI, Ma XC, Singh HB, Bi X, Pasloske BL, Howard RJ. Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36: conserved function with variant sequence. Blood 1997;90(9):3766-3775. https://doi.org/10.1182/blood.V90.9.3766
- 9. Chen Q, Barragan A, Fernandez V, Sundström A, Schlichtherle M, et al. Identification of Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) as the rosetting ligand of the malaria parasite P. falciparum.. J Exp Med 1998;187(1):15-23. https://doi.org/10.1084/jem.187.1.15
- 10. Smith JD, Subramanian G, Gamain B, Baruch DI, Miller LH. Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Mol Biochem Parasitol 2000;110(2):293-310. https://doi.org/10.1016/s0166-6851(00)00279-6
- 11. Cui L, Yan G, Sattabongkot J, Cao Y, Chen B, et al. Malaria in the Greater Mekong Subregion: heterogeneity and complexity. Acta Trop 2012;121(3):227-239. https://doi.org/10.1016/j.actatropica.2011.02.016
- 12. Parker DM, Carrara VI, Pukrittayakamee S, McGready R, Nosten FH. Malaria ecology along the Thailand-Myanmar border. Malar J 2015;14:388. https://doi.org/10.1186/s12936-015-0921-y
- 13. Ghinai I, Cook J, Hla TT, Htet HM, Hall T, et al. Malaria epidemiology in central Myanmar: identification of a multi-species asymptomatic reservoir of infection. Malar J 2017;16(1):16. https://doi.org/10.1186/s12936-016-1651-5
- 14. Zhang Q, Zhang Y, Huang Y, Xue X, Yan H, et al. From in vivo to in vitro: dynamic analysis of Plasmodium falciparum var gene expression patterns of patient isolates during adaptation to culture. PLoS One 2011;6(6):e20591. https://doi.org/10.1371/journal.pone.0020591
- 15. Sirisabhabhorn K, Chaijaroenkul W, Muhamad P, Na-Bangchang K. Genetic diversity and distribution patterns of PfEMP1 in Plasmodium falciparum isolates along the Thai-Myanmar border. Parasitol Int 2021;84:102397. https://doi.org/10.1016/j.parint.2021.102397
- 16. Kang JM, Cho PY, Moe M, Lee J, Jun H, et al. Comparison of the diagnostic performance of microscopic examination with nested polymerase chain reaction for optimum malaria diagnosis in Upper Myanmar. Malar J 2017;16(1):119. https://doi.org/10.1186/s12936-017-1765-4
- 17. Tessema SK, Nakajima R, Jasinskas A, Monk SL, Lekieffre L, et al. Protective immunity against severe malaria in children is associated with a limited repertoire of antibodies to conserved PfEMP1 variants. Cell Host Microbe 2019;26(5):579-590e5. https://doi.org/10.1016/j.chom.2019.10.012
- 18. Chen DS, Barry AE, Leliwa-Sytek A, Smith TA, Peterson I, et al. A molecular epidemiological study of var gene diversity to characterize the reservoir of Plasmodium falciparum in humans in Africa. PLoS One 2011;6(2):e16629. https://doi.org/10.1371/journal.pone.0016629
- 19. Saita S, Pan-Ngum W, Phuanukoonnon S, Sriwichai P, Silawan T, et al. Human population movement and behavioural patterns in malaria hotspots on the Thai-Myanmar border: implications for malaria elimination. Malar J 2019;18(1):64. https://doi.org/10.1186/s12936-019-2704-3
- 20. Naw H, Kang JM, Moe M, Lee J, Lê HG, et al. Temporal changes in the genetic diversity of Plasmodium vivax merozoite surface protein-1 in Myanmar. Pathogens 2021;10(8):916. https://doi.org/10.3390/pathogens10080916
- 21. Lê HG, Thái TL, Kang JM, Lee J, Moe M, et al. Genetic polymorphism of merozoite surface protein-3 in Myanmar Plasmodium falciparum field isolates. Malar J 2020;19(1):184. https://doi.org/10.1186/s12936-020-03256-y
- 22. Lê HG, Kang JM, Jun H, Lee J, Thái TL, et al. Changing pattern of the genetic diversities of Plasmodium falciparum merozoite surface protein-1 and merozoite surface protein-2 in Myanmar isolates. Malar J 2019;18(1):241. https://doi.org/10.1186/s12936-019-2879-7
- 23. Rottmann M, Lavstsen T, Mugasa JP, Kaestli M, Jensen ATR, et al. Differential expression of var gene groups is associated with morbidity caused by Plasmodium falciparum infection in Tanzanian children. Infect immun 2006;74(7):3904-3911. https://doi.org/10.1128/IAI.02073-05
- 24. Tessema SK, Monk SL, Schultz MB, Tavul L, Reeder JC, et al. Phylogeography of var gene repertoires reveals fine-scale geospatial clustering of Plasmodium falciparum populations in a highly endemic area. Mol Ecol 2015;24(2):484-497. https://doi.org/10.1111/mec.13033
- 25. Aguiar JC, Albrecht GR, Cegielski P, Greenwood BM, Jensen JB, et al. Agglutination of Plasmodium falciparum-infected erythrocytes from east and west African isolates by human sera from distant geographic regions. Am J Trop Med Hyg 1992;47(5):621-632. https://doi.org/10.4269/ajtmh.1992.47.621
- 26. Rask TS, Hansen DA, Theander TG, Gorm Pedersen A, Lavstsen T. Plasmodium falciparum erythrocyte membrane protein 1 diversity in seven genomes--divide and conquer. PLoS Comput Biol 2010;6(9):e1000933. https://doi.org/10.1371/journal.pcbi.1000933
- 27. Lavstsen T, Salanti A, Jensen AT, Arnot DE, Theander TG. Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J 2003;2:27. https://doi.org/10.1186/1475-2875-2-27
- 28. Cham GK, Turner L, Lusingu J, Vestergaard L, Mmbando BP, et al. Sequential, ordered acquisition of antibodies to Plasmodium falciparum erythrocyte membrane protein 1 domains. J Immunol 2009;183(5):3356-3363. https://doi.org/10.4049/jimmunol.0901331
- 29. Fowler EV, Peters JM, Gatton ML, Chen N, Cheng Q. Genetic diversity of the DBLalpha region in Plasmodium falciparum var genes among Asia-Pacific isolates. Mol Biochem Parasitol 2002;120(1):117-126. https://doi.org/10.1016/s0166-6851(01)00443-1
- 30. Bull PC, Berriman M, Kyes S, Quail MA, Hall N, et al. Plasmodium falciparum variant surface antigen expression patterns during malaria. PLoS Pathog 2005;1(3):e26. https://doi.org/10.1371/journal.ppat.0010026
- 31. Ozarkar A, Prakash D, Deobagkar D, Deobagkar D. Analysis of PfEMP1-var gene sequences in different Plasmodium falciparum malarial parasites. Sch Res Exch 2009;2009:1-10. https://doi.org/10.3814/2009/824949
- 32. Barry AE, Leliwa-Sytek A, Tavul L, Imrie H, Migot-Nabias F, et al. Population genomics of the immune evasion (var) genes of Plasmodium falciparum. PLoS Pathog 2007;3(5):e34. https://doi.org/10.1371/journal.ppat.0030034

 , Sanghyun Lee2,†
, Sanghyun Lee2,†