Establishing a Cre/loxP-based genetic manipulation system for Acanthamoeba: Targeted genome editing and stable reporter expression
Article information

Abstract
Acanthamoeba is an opportunistic pathogen responsible for granulomatous amoebic encephalitis and amoebic keratitis. Despite its clinical significance, effective treatments remain challenging due to a limited understanding of its pathogenic mechanism. This study developed a genetic manipulation system in Acanthamoeba to facilitate gene function and drug screening studies. We applied the Cre/loxP system to integrate the gene encoding the tdTomato fluorescent protein into the genome of Acanthamoeba castellanii via homologous recombination. The polyubiquitin gene and its untranslated regions were identified and verified, after which the tdTomato gene was cloned between the untranslated regions of the polyubiquitin gene. The construct was then introduced into the Acanthamoeba genome using a modified pLPBLP vector containing loxP sites. Cre recombinase was utilized to remove the neomycin resistance cassette flanked by loxP sites, and genetically modified cells were selected by clonal dilution. The integration of the tdTomato gene, confirmed through PCR and fluorescence microscopy, showed stable expression in both trophozoites and cysts without the need for antibiotic selection. We demonstrated the feasibility of antibiotic-free reporter gene expression in Acanthamoeba. The system provides a valuable tool for functional genomics, allowing us to explore gene functions in Acanthamoeba and develop reliable drug screening models. Furthermore, the ability to express genes without the continuous use of selection markers opens up new possibilities for studying the pathobiology of this pathogen and advancing the development of novel therapeutic strategies against Acanthamoeba infections.
Introduction
Acanthamoeba, an opportunistic human pathogen responsible for granulomatous amoebic encephalitis, dermatitis, and amoebic keratitis, is commonly found in a wide range of natural and artificial environments [1,2]. Despite the continued occurrence of Acanthamoeba infections, effective treatment remains challenging due to limited awareness of amoebae as pathogens [1–3]. Thus, developing effective therapeutic agents requires a thorough investigation of the factors critical to parasite proliferation and survival. Additionally, a reliable drug screening system is needed to accurately assess the cell response to treatments [4]. One such reliable approach involves integrating reporter genes, such as those encoding fluorescent proteins or luciferase, into the pathogen’s genome to monitor cell proliferation and survival [5]. Stable and consistent expression of these reporter genes allows for precise and long-term tracking of cellular processes. However, research progress has been hindered in Acanthamoeba due to the limited availability of genetic modifications, a shortage of reliable antibiotic selection markers and promoters, and uncertainties regarding the feasibility of homologous recombination.
The Cre/loxP system has become a powerful tool for controlling genetic manipulations in various organisms through homologous recombination, including protozoan parasites such as Dictyostelium discoideum, Giardia lambria, Toxoplasma gondii, and Plasmodium falciparum [6–11]. LoxP sequences, which are 34 bp sequences with an asymmetrical core, are initially inserted into the genome on either side of the gene of interest using homologous recombination. Cre recombinase then facilitates DNA recombination at specific loxP sites [12,13]. Depending on the orientation of these loxP sites, Cre can induce excision, insertion, inversion, or translocation of DNA. Thus, the Cre-loxP system enables site-specific recombination, allowing for precise editing of genes at designated loci. This capability is particularly advantageous for generating cells that express reporter proteins or conditional mutants, allowing the study of essential genes without compromising the parasite’s viability during crucial developmental stages.
Various studies have recently increased our knowledge of the Acanthamoeba castellanii Neff strain genome, establishing it as a foundational research model for Acanthamoeba [14–16]. Advancing research on the essential survival and virulence factors in this cell requires developing genetic manipulation techniques to establish a drug screening system to identify drug targets and evaluate potential new compounds.
In this report, we address the challenge of manipulating the A. castellanii genome by adapting the Cre/loxP system. First, the 5′- and 3′-untranslated regions (UTRs) of the polyubiquitin (pUb) gene in the A. castellanii Neff strain were screened. Then, we replaced the pUb gene with the gene encoding the tdTomato fluorescent protein and integrated it into the genome by homologous recombination. Subsequently, the antibiotic selection gene, located between the loxP sequences, was excised using Cre recombinase. This modification facilitated stable expression of the tdTomato fluorescent protein in Acanthamoeba trophozoites and cysts without the need for antibiotics.
Materials and Methods
Ethics statement
Not applicable.
Cultivation of amoebae and induction of encystation
The A. castellanii Neff strain (ATCC 30010) was acquired from the American Type Culture Collection (Manassas, VA, USA). The amoebae were cultured axenically in peptone-yeast-glucose (PYG) medium at 25°C, and encystment was induced using previously described methods [17]. Briefly, cells from cultures in the post-logarithmic growth phase were aseptically collected. Approximately 5×105 cells were washed once with phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4) and then incubated for 72 h in 10 ml of encystment medium containing 95 mM NaCl, 5 mM KCl, 8 mM MgSO4, 0.4 mM CaCl2, 1 mM NaHCO3, and 20 mM Tris–HCl (pH 9.0).
Confirmation of the pUb gene and 5′- and 3′-UTR regions in A. castellanii Neff strain
The pUb gene of the Acanthamoeba isolate 4B3 [18], along with the 5′ and 3′ UTRs from the A. castellanii Neff strain, were identified through BLAST searches in the NCBI GenBank (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and AmoebaDB (https://amoebadb.org/amoeba/app). The sequences were verified by PCR and direct sequencing (Macrogen, Seoul, Korea), and then aligned using ClustalW.
DNA construction of the Acanthamoeba Cre-loxP system, transfection, and clonal selection via limiting dilution
All oligonucleotide sequences used in this work are listed in Supplementary Table S1. The Acanthamoeba Cre-loxP system used the previously described pLPBLP expression vector from D. discoideum, which had been employed for gene disruption utilizing the Cre-loxP system (NBRP Nenkin, https://nenkin.nbrp.jp/) [8]. Initially, the floxed-blasticidin S (BSD) cassette, including the Dictyostelium promoter, was excised from the pLPBLP vector using XbaI and EcoRV restriction enzymes. Subsequently, the neomycin (NEO) cassette from the pGAPDH-EGFP-NEO vector [19], which includes a TATA binding promoter and a poly A signal, was PCR amplified using NEO-XbaI-F(forward) and NEO-EcoRV-R(reverse) primers. This was followed by digestion with XbaI and EcoRV and ligation into the 2 loxP sites of the pLPBLP vector treated with XbaI and EcoRV, thus cloning the construct (pLP-Neo-LP).
Acanthamoeba genomic DNA was used as a template to amplify the 5′-UTR and 3′-UTR of the pUb gene using 5′UTR-KpnI-F and 5′UTR-HindIII-R primers, and 3′UTR-XhoI-F and 3′UTR-SpeI-R primers, respectively. The tdTomato gene was amplified from the pCSCMV:tdTomato vector [20] using tdTomato-HindIII-F and tdTomato-XmaI-R primers. The floxed-NEO cassette was generated from the pLP-Neo-LP plasmid using Neo-XmaI-F and Neo-XhoI-R primers by PCR. All 4 amplified PCR products were ligated into the KpnI and SpeI-digested pLPBLP plasmid using the Gibson Assembly Cloning Kit (New England Biolabs, Ipswich, MA, USA). The sequences of the resulting plasmid, pUb-Tdtomato-pLP-Neo-LP, were confirmed by direct sequencing (Macrogen).
The resulting vector was then linearized with KpnI and SpeI and transfected into the A. castellanii Neff strain as previously described, using PolyFect (Qiagen, Hilden, Germany) instead of SuperFect transfection reagent (Qiagen). After 24 h of incubation, the transfected cells were transferred to a 75 cm2 cell culture flask containing 10 mL of PYG medium supplemented with G418 (final concentration 100 μg/ml, InvivoGen, San Diego, CA, USA) and incubated at 25°C for 10–14 day. Subsequently, the selected trophozoites were counted and cloned into single isolates by a limiting dilution, inoculating 96-well plates with 1 trophozoite per well in PYG medium containing the same concentration of G418 for an additional 10–14 days. Following this, genomic DNA was extracted from the clones exhibiting tdTomato expression, which was confirmed via fluorescence microscopy, and chromosome integration was verified by PCR as previously described [8]. Integration of tdTomato into the pUb gene in the A. castellanii genome was confirmed by PCR using one primer located within the tdTomato gene (tdTomato-2) and another positioned outside the 5′-UTR targeting fragment (tdTomato-1) (Supplementary Table S2). A separate PCR was performed to examine the presence of the NEO cassette using primers (NEO-3 and NEO-4) flanking the floxed-NEO insertion site.
Crystal violet assay for assessing Acanthamoeba viability after hygromycin treatment
A total of 5×10⁴ A. castellanii trophozoites in 250 μl of PYG medium were plated in 96-well plates, followed by the addition of 50 μl of various concentrations of hygromycin in PBS. The samples were incubated at 25°C for 48 h. Trophozoites cultured without hygromycin served as the control. All assays were performed in triplicate. After 48 h, cell viability was assessed using the crystal violet method as previously described [21]. Cells were washed with PBS, fixed with 100 μl of methanol for 30 sec, and stained with 100 μl of 0.1% crystal violet solution for 15 min at room temperature in the dark. After 3 saline washes, 100 μl of 1% SDS in 50% ethanol was added to solubilize the stain, and absorbance was measured at 620 nm using a Gemini EM microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Construction of an Acanthamoeba vector expressing Cre recombinase with a hygromycin selection marker
To construct a plasmid for expression of Cre recombinase in Acanthamoeba, we first amplified the Cre recombinase gene with the NLS-Cre-SpeI-F primer containing an N-terminally fused SV40 nuclear localization signal sequence and NLS-Cre-SpeI-R, with pCre-ERT2 as the template [22]. The PCR product was then digested with SpeI and ligated into the SpeI-treated pGAPDH-EGFP-NEO plasmid, generating the pGAPDH-Cre-NEO plasmid. To replace the NEO cassette in pGAPDH-Cre-NEO with a hygromycin resistance gene, the plasmid was first digested with AflII and BstBI to remove the NEO cassette. The HYG gene was then amplified from the pIREShyg3 (Clontech, CA, USA) plasmid using the Hyg-AflII-F and Hyg-BstBI-R primers, digested with AflII and BstBI, and ligated into the AflII- and BstBI-treated plasmid, generating the final construct (pGAPDH-Cre-HYG).
Transfection, clonal selection, and validation of Cre-loxP mediated recombination in Acanthamoeba
Clones with confirmed integration of the tdTomato gene and the floxed-NEO cassette into the Acanthamoeba genome were transfected with the pGAPDH-Cre-HYG vector, which was not linearized, following the transfection method described above. After 24 h of incubation, hygromycin (final concentration 100 μg/ml, InvivoGen) was added, and clonal selection was performed by limiting dilution, as described above. The clonal cell lines were then picked and replicated on 2 different 96-well plates. One plate was treated with G418 (final concentration 100 μg/ml), while the other plate continued to be treated with HYG (final concentration 100 μg/ml) for 7–10 days. At every stage, the A. castellanii Neff strain with no vector transfection and empty vector transfection served as the control for the wild-type parental cell line. Cells from the positions in the replica plates treated with HYG, corresponding to those not growing in G418 in the 96-well plates, were selected and plated on 24-well plates containing media without G418 or hygromycin for further culture. After 21–28 days of continuous cultivation without antibiotics, the cells were replica plated again, treated with HYG to select only HYG-sensitive cells, and then genomic DNA was extracted from these G418- and hygromycin-sensitive cells. The absence of the floxed-NEO cassette and HYG cassette was verified by PCR using NEO-3 and NEO-4 primers and HYG-5 and HYG-6 primers (Supplementary Table S2), respectively. For clonal cells where Cre recombination was confirmed, the expression of tdTomato fluorescent protein was assessed in trophozoites and cysts using an LSM5 Exciter scalable confocal microscope (Zeiss, Hamburg, Germany). TdTomato-mediated fluorescence was detected using a band-pass filter with an excitation wavelength of 554 nm and an emission wavelength of 581 nm. Cell images were acquired and analyzed using a Zeiss LSM Image Examiner (Zeiss).
Results
Construction of the floxed-NEO targeting vector
To integrate and express exogenous genes in the A. castellanii genome through homologous recombination, we used the pUb gene from the Acanthamoeba isolate 4B3 and its promoter sequence (GenBank accession No. X87156) [18], which had been previously used in the pUb vector [23], to screen the 5′-UTR and 3′-UTR sequences of the pUb gene in the A. castellanii Neff strain genome database. Given the potentially significant differences in the 5′-UTR and 3-UTR sequences of the pUb gene among Acanthamoeba species, we first searched for the pUb gene corresponding to the reported X87156 in the A. castellanii Neff genome database and identified a transcript with 100% amino acid identity (Supplementary Fig. S1) and 92.3% nucleotide sequence identity (Fig. 1) to the coding region of the X87156, which had been annotated as pUb (GenBank accession No. ELR19129). The 5′-UTR regions of ELR19129 from the A. castellanii Neff strain genomic scaffold sequence (GenBank accession No. KB007936) and the reported X87156 showed 51% identity (Fig. 1).

Sequence alignment of the Acanthamoeba castellanii Neff polyubiquitin coding region (GenBank accession No. ELR19129) genomic scaffold sequence containing the 5′- and 3′-UTR regions (GenBank accession No. KB007936) compared to the reported Acanthamoeba polyubiquitin gene (GenBank accession No. X87156). Three ubiquitin repeat units including ATG codons are highlighted with grey bars above the corresponding regions. Regulatory elements such as the octamer, TATA box, BREu, and CAAT box are indicated with black squares.
Approximately 500 base pairs of the 5′ and 3′-UTRs of the A. castellanii pUb coding region were PCR-amplified and cloned into the pLPBLP vector [14], which contains loxP sites and is used in D. discoideum for gene targeting and manipulation. To examine whether exogenous genes can integrate into the Acanthamoeba genome via homologous recombination, we cloned the gene encoding the tdTomato fluorescent protein between the 5′- and 3′UTRs of the pUb gene in A. castellanii. The constructed vector (pUb-tdTomato-LPBLP) was further modified by replacing the BSD resistance gene with a NEO resistance gene, serving as a selection marker in Acanthamoeba, resulting in the pUb-tdTomato-LP-Neo-LP plasmid (Fig. 2; Supplementary Fig. S2). The complete plasmid sequence and vector map are available on Addgene (No. 231509, https://www.addgene.org/).
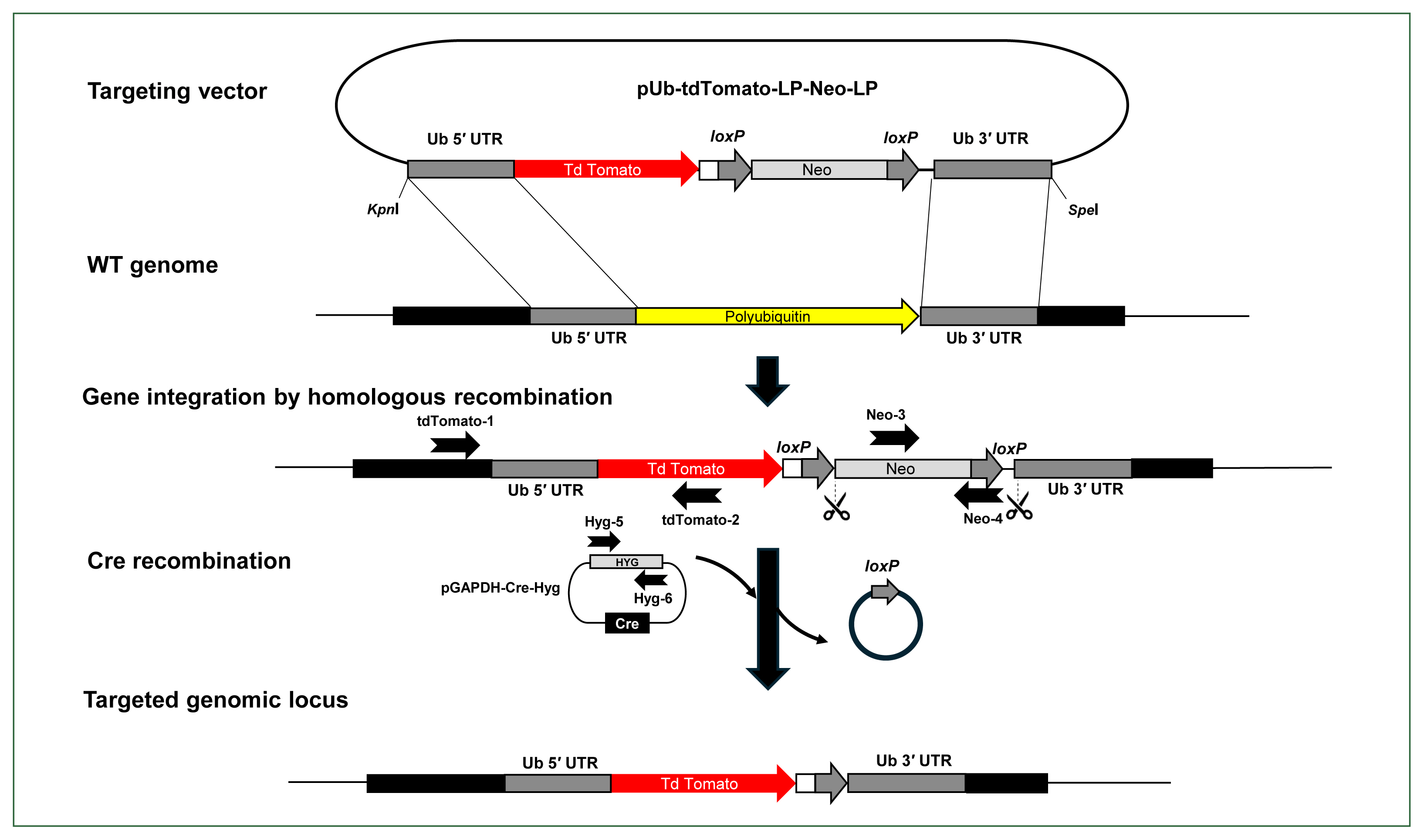
Schematic illustration of the Cre/loxP reporter system developed in Acanthamoeba castellanii. The tdTomato gene was cloned between the 5′ and 3′ UTRs of the polyubiquitin coding region and integrated into the Acanthamoeba genome using a modified pLPBLP vector with loxP sites. Neomycin resistance was used for selection, and the Cre recombinase gene was expressed using a separate plasmid with hygromycin resistance for selecting Cre-expressing cells, facilitating the excision of the loxP-Neo-loxP cassette. The right and left arrows above the gene regions represent the primers used for PCR to verify the integration of the gene encoding the tdTomato fluorescent protein into the genome (tdTomato-1 and tdTomato-2), the presence or absence of the floxed neo cassette (Neo-3 and Neo-4), and the retention of the pGAPDH-Cre-Hyg plasmid (Hyg-4 and Hyg-5). Scissors indicate the region targeted for excision by Cre recombinase. WT, wild type A. castellanii.
After integration of the linearized pUb-tdTomato-LP-Neo-LP plasmid, the floxed NEO cassette in the Acanthamoeba genome was excised by Cre recombinase. To achieve this, a new plasmid capable of expressing the Cre recombinase gene in Acanthamoeba was constructed by cloning the Cre recombinase gene into the pGAPDH-EGFP-NEO plasmid. Since Acanthamoeba transfectants with the integrated pUb-Tdtomato-LP-Neo-LP construct contained the NEO cassette, a new antibiotic resistance marker was needed to select cells expressing Cre recombinase. Among various antibiotics tested, hygromycin was identified as an appropriate selection marker for Acanthamoeba (Fig. 3A). Therefore, NEO was replaced with the hygromycin B phosphotransferase gene (HYG) in the pGAPDH-Cre-NEO plasmid, resulting in the construction of pGAPDH-Cre-HYG (Fig. 2; Supplementary Fig. S2). The complete plasmid sequence and vector map are available on Addgene (No. 231510, https://www.addgene.org/).
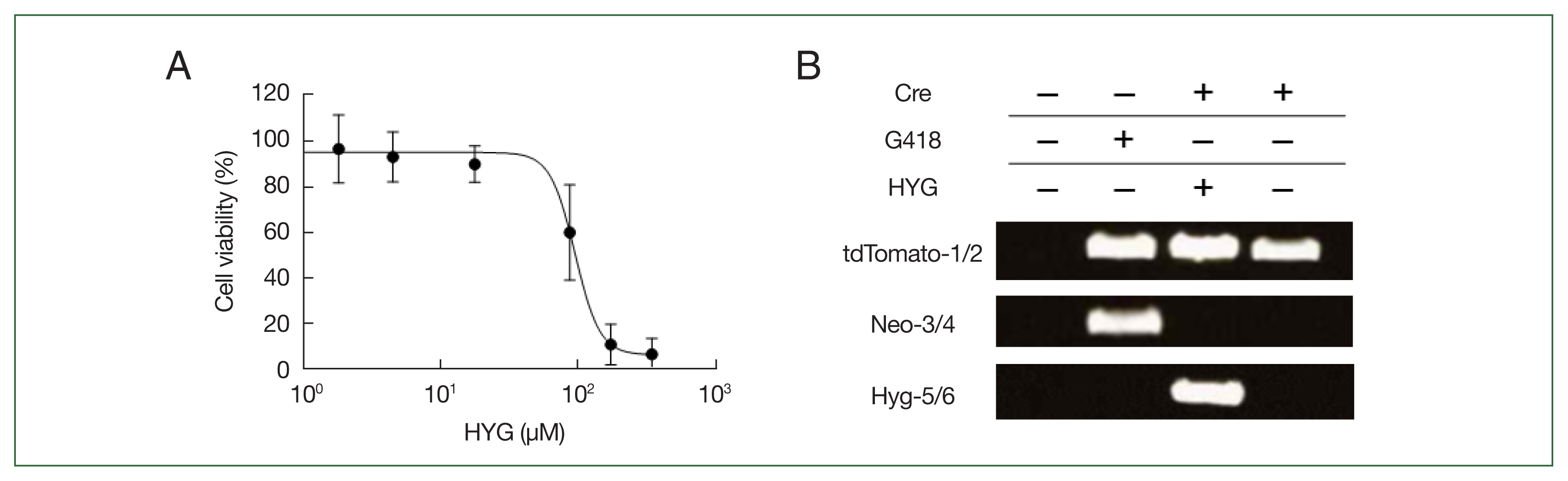
Cell viability of the Acanthamoeba castellanii in response to hygromycin (HYG) treatment and PCR validation of gene integration and Cre recombinase activity in Acanthamoeba. (A) Acanthamoeba viability was assessed using a crystal violet assay following HYG treatment. After treating A. castellanii with varying concentrations of HYG, cell density was determined by scanning of crystal violet staining at 48 h. HYG concentrations are presented on a logarithmic scale, and cell viability is expressed as the percentage of viable cells relative to untreated controls, with error bars representing the standard deviation. (B) PCR validation of tdTomato gene integration (tdTomato-1 and tdTomato-2, upper panel), Cre recombinase-mediated excision of the floxed-NEO cassette (Neo-3 and Neo-4, middle panel) from the Acanthamoeba genome, and loss of the pGAPDH-Cre-Hyg plasmid (Hyg-5 and Hyg-6, lower panel). Primer locations are shown as right and left arrows in Fig. 2. Cre (−) represents clones that were G418-resistant before transfection with pGAPDH-Cre-Hyg, while Cre (+) represents clones that became G418-sensitive after transfection and were not treated with HYG for over 2 months.
Validation of Cre-mediated recombination
After transfecting A. castellanii with the constructed pUb-tdTomato-pLP-Neo-LP plasmid, selection was performed using G418. Transfectants were subjected to limiting dilution to isolate individual colonies. Genomic DNA was then extracted from these clonal isolates to examine whether tdTomato was integrated into the target regions through homologous recombination at the 5′- and 3′-UTR of the pUb gene, confirmed by PCR with distinct primer sets. To confirm chromosomal integration of the tdTomato gene, PCR was conducted using a primer located outside the upstream region of the 5′-UTR sequence of pUb gene (tdTomato-1) and a primer within the tdTomato gene sequence (tdTomato-2) (Fig. 2; Supplementary Table S2). We also employed primers to detect the integration of the floxed NEO cassette and its subsequent excision by Cre recombinase (Neo-3 and Neo-4). After transient transfection with pGAPDH-Cre-Hyg and selection via hygromycin for Cre-loxP recombination, we utilized primers (Hyg-5 and Hyg-6) to demonstrate the loss of the pGAPDH-Cre-Hyg plasmid due to the absence of selection (Fig. 2; Supplementary Table S2). PCR products using the tdTomato-1 and tdTomato-2 primers were not detected in wild-type controls but were observed in pUb-tdTomato-LP-Neo-LP plasmid-transfected cells both before (Cre (−)) and after pGAPDH-Cre-Hyg transfection (Cre (+)) (Fig. 3B, upper), indicating that the tdTomato gene successfully replaced the pUb gene in the A. castellanii genome. PCR using the Neo-3 and Neo-4 primers targeting the floxed NEO cassette produced a band in Cre (−) cells, but this band disappeared in Cre (+) cells (Fig. 3B, middle). A. castellanii cells in which tdTomato gene integration and floxed NEO cassette removal were confirmed were continuously cultured for approximately 2 months in media without G418 and hygromycin. No PCR products were detected in the Cre (+) cells using the Hyg-5 and Hyg-6 primers for HYG, indicating that the pGAPDH-Cre-Hyg plasmid had disappeared from the tdTomato gene-integrated cells (Fig. 3B, lower). G418- and hygromycin-sensitive cells with integrated tdTomato genes were then analyzed for tdTomato expression using confocal microscopy. As shown in Fig. 4A, tdTomato fluorescence was observed in both trophozoites and cysts of A. castellanii even without G418 and hygromycin selection. Notably, centrifugation of the cells followed by visual inspection under visible light revealed a red indicative of tdTomato protein expression (Fig. 4B).
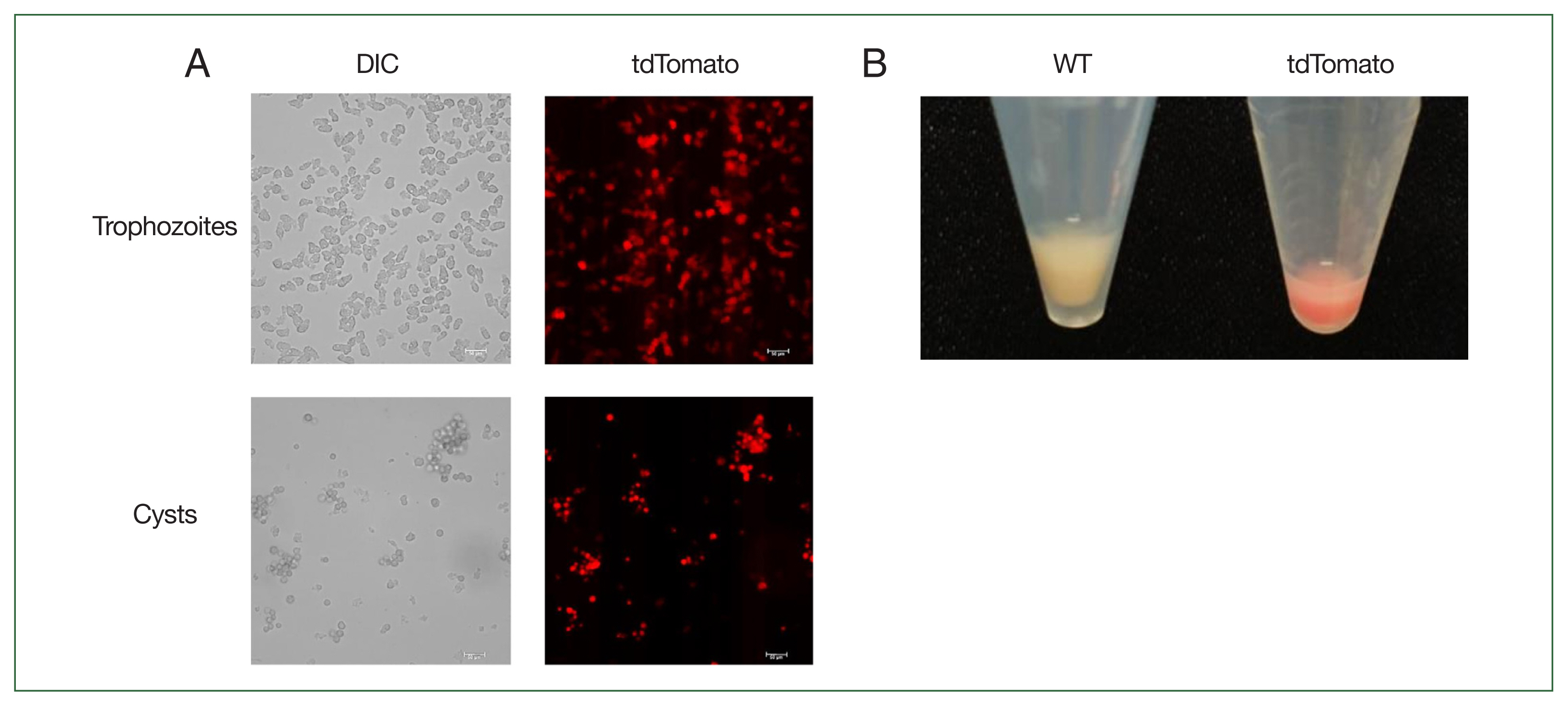
Observation of tdTomato expression in Acanthamoeba castellanii using fluorescence microscope. (A) Fluorescence microscopic observation of tdTomato expression in trophozoites and cysts. Scale bar=50 μm. (B) Visual comparison of cell pellets after centrifugation between wild type and tdTomato-expressing cells. DIC, differential interference contrast; WT, wild-type A. castellanii; tdTomato, A. castellanii with the tdTomato gene integrated into the genome, sensitive to both G418 and hygromycin.
Discussion
In Acanthamoeba, systems for expressing exogenous genes or overexpressing specific endogenous genes remain limited, primarily due to the scarcity of effective promoters—especially those capable of driving protein expression in cysts—as well as the lack of reliable drug resistance markers for selecting transfectants and the overall low transfection efficiency. Promoters reported to date include those for TATA binding promoter [24], transcription factor IID [25], cyst-specific protein 21 [26], glyceraldehyde phosphate dehydrogenase [19], and pUb from the Acanthamoeba isolate 4B3 [18].
In this study, we identified the orthologue of the pUb gene from Acanthamoeba isolate 4B3 in the A. castellanii Neff strain and characterized the 5′-UTR, containing the promoter, as well as the 3′-UTR of the pUb gene in A. castellanii to facilitate gene expression and manipulation through homologous recombination. The pUb gene (X87156) from Acanthamoeba isolate 4B3 consists of a 515-nt coding region, which includes 2 complete ubiquitin repeat units and one partial ubiquitin repeat unit, along with a 1,873 bp 5′ upstream region (Fig. 1). PCR and sequencing analysis confirmed that the pUb gene from A. castellanii is identical to the sequence annotated as pUb in the genome database (GenBank accession No. ELR19129). ELR19129 comprises 3 ubiquitin repeat units, with an additional asparagine (N) at the end of the third repeat unit followed by a stop codon (Fig. 1; Supplementary Fig. S1). Although the pUb gene from Acanthamoeba isolate 4B3 is a partial sequence, the amino acid sequences of the ubiquitin repeat units of pUb are 100% identical to those in ELR19129. ELR19129 encodes a protein of 229 amino acids with an expected molecular weight of 25.9 kDa. The 5′-UTR of KB007936, the genomic scaffold sequence that includes ELR19129, contains key conserved putative regulatory elements also found in the 5′-UTR of X87156, such as an Octamer [27,28], a CAAT box [29], and a TATA box [25,30] (Fig. 1). Additionally, the B recognition element (GGGCGCC or CCACGCC), located immediately upstream of the TATA box and responsible for binding transcription factor IIB to facilitate RNA polymerase II binding and regulate transcription levels, was also found to be conserved [31,32]. In other organisms, ubiquitin promoters ensure strong, constitutive expression across developmental stages, resist silencing, and maintain stable expression under stress, making them versatile and reliable for transgene expression [33–35]. Due to their conserved regulatory elements, pUb promoters are also considered ideal for overexpressing foreign genes or specific proteins in Acanthamoeba. As expected, tdTomato expression was consistently maintained not only in actively proliferating trophozoites but also in dormant cysts of Acanthamoeba (Fig. 4A).
The drug-resistance genes that allow for the selection of successfully transformed Acanthamoeba cells are limited. In this study, the Dictyostelium pLPBLP vector, which contains a BSD cassette gene between 2 loxP sites, was initially used. However, blasticidin selection in Acanthamoeba proved ineffective (data not shown), so the BSD cassette was replaced with the NEO gene for transformant selection. Next, to facilitate the removal of the floxed NEO cassette integrated into the Acanthamoeba chromosome via Cre recombinase expression, a new antibiotic resistance gene available in Acanthamoeba was required as a selection marker. Various concentrations of hygromycin were applied to Acanthamoeba trophozoites, and cell viability was measured after 48 h using a crystal violet assay, determining the IC50 of hygromycin to be 98.68 μM (51.1 μg/mL, R2=0.9253) (Fig. 3A). In Acanthamoeba transfected with the pGAPDH-Cre-Hyg plasmid, the floxed NEO cassette was excised from the genome with an efficiency of 14.6%±4.3%. In these clonal isolates, plasmid loss of pGAPDH-Cre-Hyg occurred when hygromycin was no longer added, and these cells subsequently became sensitive to both NEO and hygromycin. Therefore, in cases where the target gene is non-essential for cell survival, repeated application of the Cre-loxP system, utilizing the floxed NEO cassette and a Cre recombinase plasmid containing HYG, is expected to facilitate gene knockout in Acanthamoeba, similar to its successful use in other protozoa [8,9,36]. In this study, stable integration of the gene encoding tdTomato enabled continuous and high-level expression of this fluorescent protein without the need for antibiotics (Fig. 4). Thus, using the Cre-loxP system to construct Acanthamoeba strains with stable and high expression of reporter genes, such as luciferase or fluorescent proteins, would provide a valuable system for monitoring cell proliferation. Additionally, this system could be adopted as a tool for high-throughput screening in drug discovery. Taken together, we have successfully developed the stable cell line in Acanthamoeba using the Cre-loxP system to precisely integrate foreign genes into specific locations in the Acanthamoeba genome through homologous recombination. Our findings also demonstrate the potential utility of this system for gene manipulation in Acanthamoeba, where molecular biology tools remain limited.
Notes
Author contributions
Conceptualization: Hong Y
Formal analysis: Aung JM, Joo SY
Funding acquisition: Hong Y
Investigation: Aung JM, Joo SY, Bang S, Shin M
Methodology: Aung JM, Joo SY, Bang S
Resources: Aung JM, Joo S, Bang S
Supervision: Na BK, Shin M, Goo YK, Hong Y
Visualization: Shin M, Goo YK
Writing – original draft: Na BK, Shin M, Goo YK, Hong Y
Writing – review & editing: Hong Y
Conflict of interest
The authors declare no conflict of interest related to this study.
Acknowledgments
This work was supported by the National Research Foundation of Korea grant funded by the Korea government (NRF-2022R1A2C1005181).
Supplementary Information
Supplementary material is available with this article at https//doi.org/10.3347/PHD.24078.