Complete mitogenome sequence of Caryophyllaeus brachycollis (Cestoda: Caryophyllidae) from China: Characterization and phylogenetic analyses of Caryophyllidea
Article information

Abstract
Caryophyllaeus brachycollis mainly parasitizes the intestines of globally distributed freshwater fishes, and infection causes significant economic losses to the aquaculture industry. However, data on the molecular epidemiology, population genetics, and systematics of C. brachycollis are scarce. In this study, we sequenced the complete mitogenome of C. brachycollis isolated from Beijing, China. This circular mitogenome comprised 14,273 bp, which was 231 bp shorter than that of C. brachycollis isolated from Wuhan, China. The mitogenome contained 12 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and 2 noncoding regions. Bayesian inference revealed that C. brachycollis belonged to the family Caryophyllaeidae. The taxonomic status of C. brachycollis is controversial when based solely on morphological features. A comparative analysis of the mitogenome sequence obtained in this study revealed novel molecular markers for the accurate ascertainment of the phylogenetic position of this parasite.
Introduction
Caryophyllideans, the earliest diverging group of non-segmented tapeworms, are described by morphological and biological features and typically have a monozoic (non-proglottized) body plan [1–3]. They are globally distributed and host-specific, mainly parasitizing in the intestinal tracts of Cypriniformes and Siluriformes fishes. Infection leads to weight loss, reduced fecundity, and high mortality rates, severely impacting the aquaculture industry [4–9]. The evolution of Eucestoda is unclear. The results of studies analyzing nuclear DNA genes (large and small subunits of ribosomal DNA) suggest that Spathebothriidea is the most divergent eucestode [10,11], whereas those analyzing morphological features and large fragments of mitochondrial DNA suggest that Caryophyllidea is the earliest divergent taxon [3,12]. Therefore, a better understanding of the relationship between orders Spathebothriidea and Caryophyllidea is warranted.
Although the order Caryophyllidea is regarded as a key group in Eucestoda research, the systematic classification and phylogenetic relationships of its members remain controversial. The families Balanotaeniidae, Capingentidae, Caryophyllaeidae, and Lytocestidae [1,2] are generally considered to belong to this order. Caryophyllaeidae, which frequently parasitize cyprinid fish in the Palearctic region, is the biggest group within Caryophyllidea, comprising 20 genera [13]. However, a recent study suggested reclassification into 10 genera [2], including Caryophyllaeus (Gmelin, 1790). The number of species in this genus varies from the previously described 42 nominal taxa to the recently recognized 7 valid species [14,15], of which Caryophyllaeus brachycollis is an important member. C. brachycollis consists of 2 morphotypes based on morphological and molecular (large subunit of ribosomal DNA and cox1) characteristics [16,17]. Although the mitogenome is a useful marker for tapeworm identification, differentiation, and systematic analyses [18–21], the complete mitogenome is only available for 1 morphotype, which was isolated from Wuhan, Hubei Province, China (CBCW). Because there is no data on the other morphotype, the molecular differentiation of C. brachycollis in China is unclear.
Thus, our study focused on obtaining the complete mitogenome of the other C. brachycollis morphotype, which was isolated from Beijing, China (CBCB). The aims were as follows: (i) sequence and annotate the complete CBCB mitogenome, (ii) determine differences between CBCB and CBCW, (iii) infer the phylogenetic relationship among members of the order Caryophyllidea, and (iv) identify the systematic and taxonomic status of the genus Caryophyllaeus. These findings would provide more molecular data for use in further Caryophyllidea analyses.
Materials and Methods
Ethics statement
The experimental protocol was established according to the ethical guidelines of the Institutional Animal Care and Use Committee and approved by the Ethics Committee of Hunan Agricultural University (No. 202282).
Sample collection and DNA extraction
Samples were collected from the intestines of Cyprinus carpio from Beijing Zoo, China. They were washed in physiological saline, and those morphologically identified as caryophyllidean were fixed in 70% (v/v) ethanol and stored at −40°C until use.
Total genomic DNA was extracted using sodium dodecyl sulfate/proteinase K treatment, followed by spin-column purification (Wizard SV Genomic DNA Purification System; Promega, Madison, WI, USA). The purified DNA was used as the template in PCR assays with the primer pair JB3/JB4.5 targeting a fragment of cox1 for amplification [22].
Sequencing, assembling, and verification
A genomic DNA library (with 350 bp inserts) was prepared and sequenced by Novogene (Tianjin, China) using a HiSeq 2500 sequencing platform (Illumina, San Diego, CA, USA) to obtain 250 bp paired-end reads. Geneious v11.1.5 software was used to filter and clean the raw data and assemble the complete CBCB mitogenome using the amplified cox1 sequences as initial references. The parameters were set as follows: minimum overlap identity, 99.0%; minimum overlap, 150 bp; and maximal gap size, 5 bp. The assembled mitogenome was further verified by long PCR experiments (Supplementary Fig. S1) using the primers listed in Supplementary Table S1.
Annotation, visualization, and sequence analysis
The boundaries of protein-coding genes (PCGs) were identified using the NCBI tool ORF Finder (https://www.ncbi.nlm.nih.gov/orffinder/). The initiation/termination codons for each PCG were identified following alignment with the CBCW mitogenome sequence using MAFFT v7.122 software. All PCG nucleotide sequences were translated to amino acid sequences using MEGA v11.0. Twenty-two transfer RNA (tRNA) genes were detected using ARWEN [23] and tRNAscan-SE [24]. Nucleotide composition and codon usage frequencies of the PCGs were analyzed using MEGA v11.0. The annotated CBCB mitogenome was visualized using the online server Proksee (https://proksee.ca/). The CBCW sequence was used as the reference genome for comparisons with CBCB.
Phylogenetic analyses
Phylogenetic analyses were performed using 6 mitogenome sequences of tapeworms within the order Caryophyllidea (Table 1) and Paruterina candelabraria (GenBank accession No. NC_039533) as the outgroup. Amino acid sequences were aligned using MAFFT v7.122 and concatenated into a single dataset. Ambiguous bases or gaps were excluded using Gblocks 0.91b software with the default parameters [25]. Phylogenetic analyses were performed using Bayesian inference (BI) and maximum likelihood (ML) methods. BI analysis was run in MrBayes 3.1.1 using an approach similar to that of Ronquist and Huelsenbeck [26]. ML analysis was performed using PhyML 3.1 software. “JTT+I+G+F” was selected as the best suitable model by ProtTest v3.4.2 based on the Akaike information criterion [27] and was used for the BI and ML analyses. Phylograms were drawn using FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree).
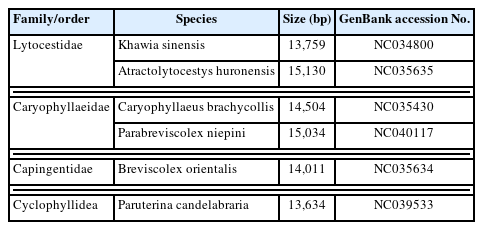
Complete mitogenome sequences of the order Caryophyllidea used for phylogenetic analysis
cox1 sequence analysis
To determine the systematic relationship of the families in Caryophyllidea, BI analysis was performed using 20 cox1 sequences of the order Caryophyllidea and Gigantolina magna (GenBank accession No. JQ268545) as the outgroup.
Results
Genome composition and organization
Sequencing of the CBCB mitogenome generated 4.08 Gb of raw data, from which approximately 7,577,818×2 clean reads were obtained following filtering and used for further assembly. The complete circular CBCB mitogenome (GenBank accession No. OP359069) was 14,273 bp and comprised 36 genes, including 12 PCGs (excluding atp8), 22 tRNA genes, 2 ribosomal RNA genes (rrnS and rrnL), and 2 noncoding regions (NCRs; control or AT-rich regions) (Fig. 1; Table 2). All 36 genes were transcribed in the forward direction. In C. brachycollis mitochondrial DNA, the AT-skews were all negative (−0.481 to −0.125), and the GC-skews were all positive (0.167 to 0.465) (Table 3), suggesting a bias toward T and G bases. The lengths of the 2 NCRs in CBCB were 845 and 423 bp, respectively, while those in CBCW were 641 and 871 bp, respectively (Table 2). The large NCR (LNCR) was located between tRNA-Leu2 and cox2, and the small NCR (SNCR) was located between tRNA-Gly and cox3 (Fig. 1). Alignment using BLAST revealed 100.0% nucleotide similarity with CBCW (GenBank accession No. NC_035430).
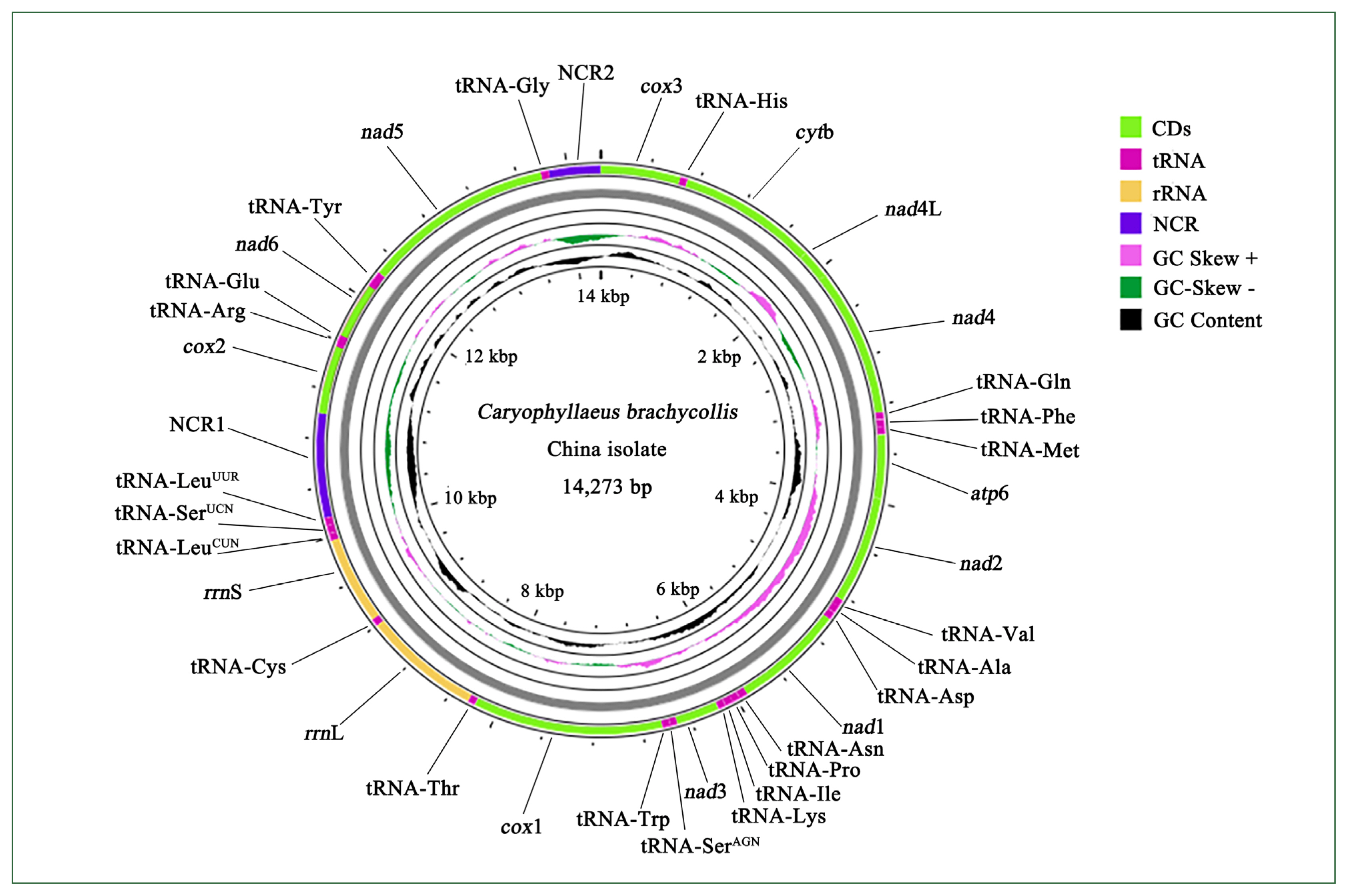
Mitogenome organization of Caryophyllaeus brachycollis isolate from China. Gene scaling is approximate. CD, coding region; NCR1, long noncoding region; NCR2, short noncoding region; tRNA, transfer RNA; rRNA, ribosomal RNA.
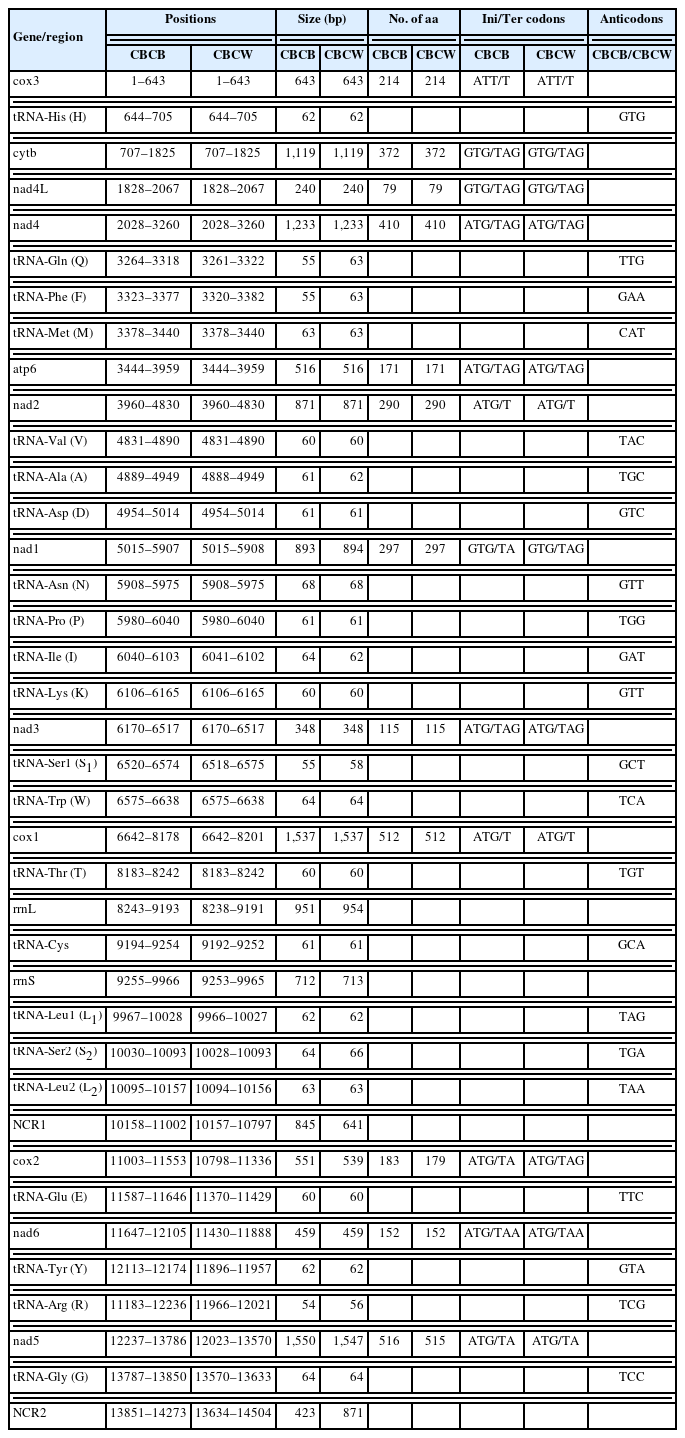
Comparative organization of the complete mitogenome of Caryophyllaeus brachycollis isolates from Beijing and Wuhan, China
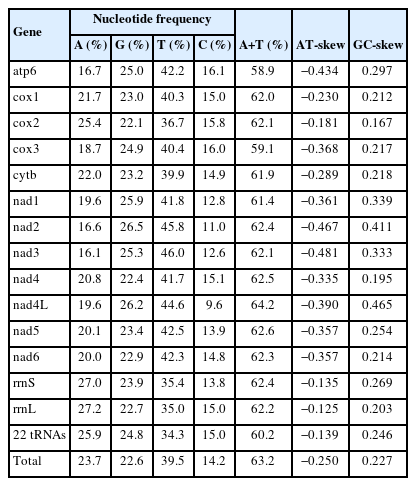
Nucleotide composition and skews of the Caryophyllaeus brachycollis mitogenome
Composition of PCGs, ribosomal RNA genes, and tRNA genes
ATN was extensively used as the initiation codon, especially ATG. GTG is an uncommon start codon in tapeworms, but it was observed in cytb, nad1, and nad4L in this study (Table 2). Furthermore, TAA and TAG were frequently used as termination codons (TAG: cytb, atp6, nad3, nad4, and nad4L; TAA: nad6) (Table 2). Incomplete stop codons T and TA were also prevalent in termination, with cox1, cox3, and nad2 using T and cox2, nad1, and nad5 using TA (Table 2). The AT-skews of the 12 protein genes were all negative (−0.481 (nad3) to −0.181 (cox2)), and the GC-skews were all positive (0.167 (cox2) to 0.465 (nad4L)), suggesting a large T base bias in nad3, a G base tendency in nad4L, and a minor tendency of T+G bases in cox2.
The 22 tRNA genes ranged from 54 to 68 bp (Table 2). rrnL and rrnS were 951 and 712 bp, respectively, with an A+T content of 62.2% and 62.4%, respectively (Table 3).
Comparative analyses of CBCB and CBCW
Comparisons between the CBCB and CBCW mitogenome sequences revealed that CBCB was approximately 231 bp shorter than CBCW. In both CBCB (Fig. 1) and CBCW, the SNCR and LNCR were located in “tRNA-Gly and cox3” and “tRNA-Leu2 and cox2,” respectively. The size (cox2 and nad5) and the initiation/termination (cox2) codons of PCGs in CBCB and CBCW differed (Table 2). cox2 was 539 bp and 551 bp in CBCW and CBCB, respectively, while nad5 was 1,547 and 1,550 bp, respectively. The initiation/termination codons of cox2 were ATG/TAG and ATG/TA in CBCW and CBCB, respectively.
Comparison of the nucleotide sequences of PCGs between CBCB and CBCW revealed differences of 0% (nad4L) to 2.2% (cox3) (Table 4). Differences in the amino acid sequences of CBCB and CBCW ranged from 0% (nad4L) to 2.7% (cox2) (Table 4). nad4L typically shows interspecies variability. However, our study revealed that it was highly conserved in C. brachycollis. These findings suggested that cox2 might be a better molecular marker for identifying cryptic species within C. brachycollis.
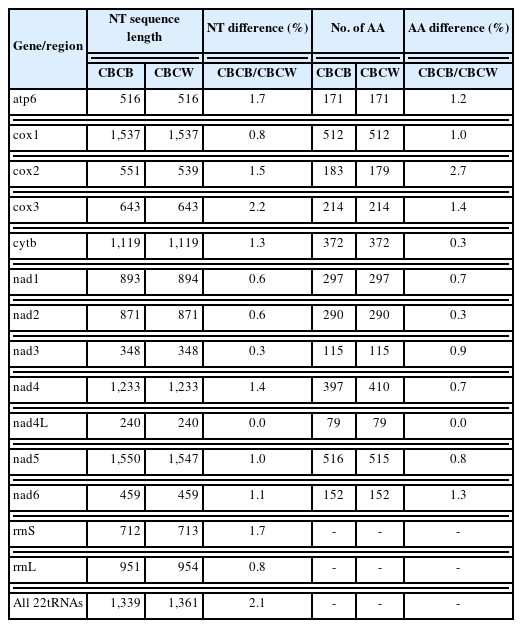
Nucleotide and amino acid sequence differences between the isolates from Beijing, China and Wuhan, China mitogenomes
Phylogenetic analyses
The phylogeny of Caryophyllidea is currently unclear due to a lack of complete mitogenome sequences. To better clarify the phylogenetic relationships among the members of this order, 6 mitogenome sequences were phylogenetically analyzed using BI and ML methods. The BI and ML tree topologies were identical (Fig. 2). There were 2 main clades: 1 clade contained Caryophyllaeidae and Lytocestidae, and the other clade contained Capingentidae, Caryophyllaeidae, and Lytocestidae, indicating that families Caryophyllaeidae and Lytocestidae were paraphyletic. The mitochondrial DNA sequences of CBCB and CBCW grouped with short topological distance (BPP =1, BF=100) (Fig. 2), indicating that they were closely related. Furthermore, phylogenetic analyses of cox1 also showed that Caryophyllaeidae and Lytocestidae were paraphyletic and revealed a close evolutionary distance between CBCB and CBCW (Supplementary Fig. S2).
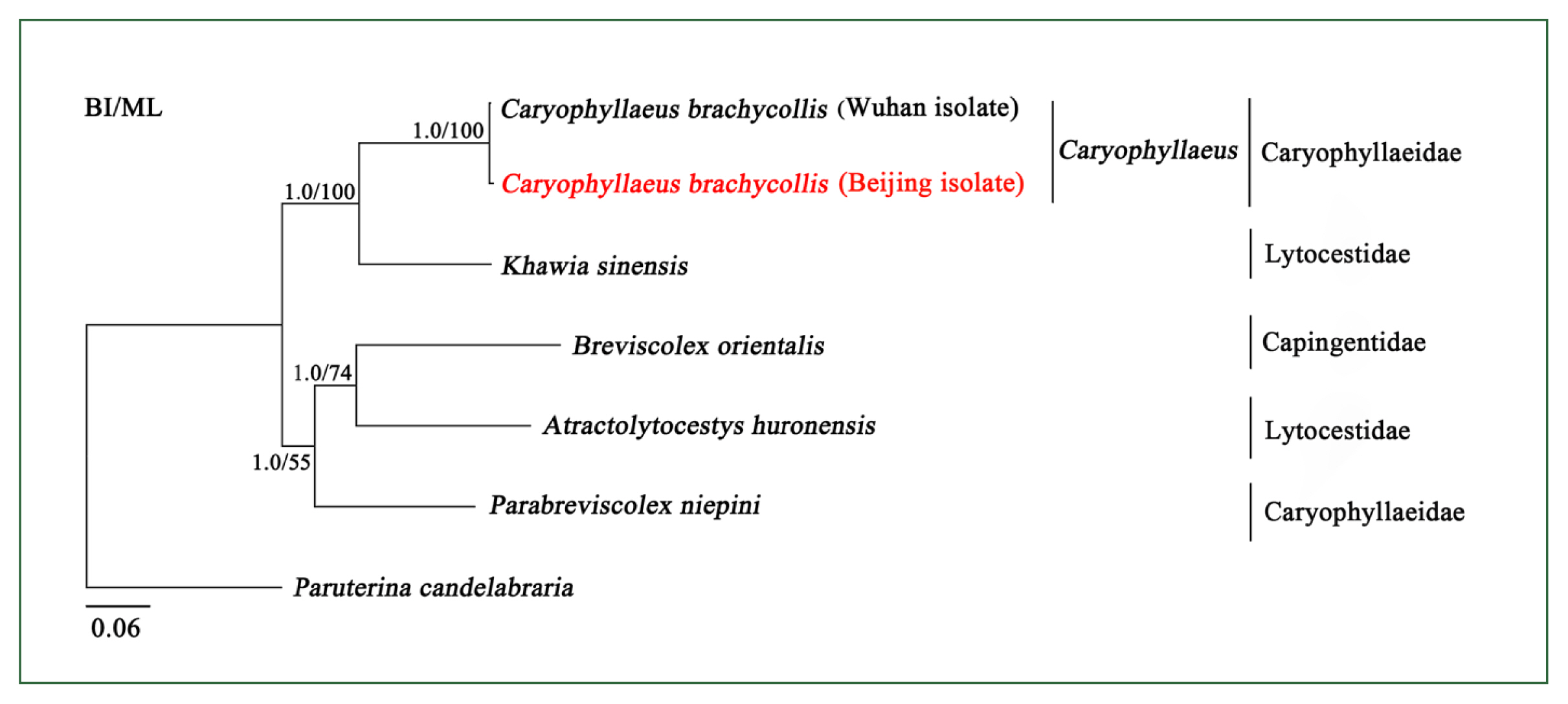
Inferred phylogenetic relationships among species in the order Caryophyllidea. The concatenated amino acid sequences of 12 mitochondrial protein-coding genes were analyzed utilizing maximum likelihood (ML) and Bayesian analysis (BI) methods, with Paruterina candelabraria as the outgroup. The Caryophyllaeus brachycollis form was isolated from carp tapeworm in Beijing in this study.
Discussion
The complete mitogenome of CBCB was 14,273 bp and encoded 36 genes. The A+T base bias (73.2%) was significantly higher than the G+C base bias (26.8%). The AT-skew ([A-T]/[A+T]) and GC-skew ([G-C]/[G+C]) were useful methods to estimate compositional asymmetry [28].
Regarding the initiation codons, ATG was the most prevalent in 8 of the 12 PCGs (atp6, cox1, cox2, nad2, nad3, nad4, nad5, and nad6), and ATT was used in cox3. TAA and TAG have been reported as common termination codons [29,30] and were identified as frequent termination codons in our study. There were 22 tRNA genes in the CBCB mitogenome. Similar to tapeworms Breviscolex orientalis and Parabreviscolex niepini [31,32], most tRNA genes in CBCB presented as a typical ‘cloverleaf’ structure, excluding tRNA-SerAGN and tRNA-Arg, which lacked DHU arms. The rrnL was located between tRNA-Thr and tRNA-Cys, and rrnS between tRNA-Cys and tRNA-Leu1, similar to the arrangement in B. orientalis and Atractolytocestus huronensis [32].
Although the mitogenome sequence of CBCW was longer than that of CBCB, the arrangements of the 36 genes were the same and consistent with the order observed in most caryophyllideans [31,32]. Considering that the differences in mitogenome sequences between closely related nematode species are 10.0%–20.0% [33,34], and species-level variations in mammals are typically 2.0%–10.0% [35], the level of differences between CBCB and CBCW indicated that they were identical.
Comparison between the CBCB and CBCW mitogenomes revealed that cox2 is a reliable molecular marker for cryptic species identification. Furthermore, nad4L was highly conserved in C. brachycollis. This comparative mitogenomic analysis elucidated evolutionary and phylogenetic relationships among related species, enhancing the accuracy of taxonomic classification.
To summarize, our study determined and characterized the complete mitogenome sequence (14,273 bp) of CBCB, revealing that it was shorter than that of CBCW. The CBCB mitogenome provided evidence supporting a close phylogenetic relationship between C. brachycollis and other members of the Caryophyllidae family. Because only a limited number of specimens from a single geographic location were analyzed, future studies should include a broader sample to further assess intraspecific variations.
Notes
Author contributions
Conceptualization: Liu YL, Fu YT
Data curation: Liu YL, Zhang Y, Fu YT
Formal analysis: Zhang Y
Investigation: Liu YL
Methodology: Deng YP
Resources: Wang HM, Deng YP
Software: Wang HM
Supervision: Fu YT, Liu GH
Validation: Zhang Y, Fu YT, Liu GH
Writing – original draft: Liu YL, Zhang Y, Wang HM
Writing – review & editing: Deng YP
Conflict of interest
The authors declare no conflict of interest related to this study.
Acknowledgments
This study was supported in part, by the Training Programme for Excellent Young Innovators of Changsha (grant No. KQ2106044). The raw data have been deposited in GenBank (accession No. OP359069 and SRR34793993).
Supplementary Information
Supplementary material is available with this article at https://doi.org/10.3347/PHD.25044.