Reconstruction of the nearly complete mitochondrial genome of a Danish isolate of Hymenolepis diminuta (Cyclophyllidea: Hymenolepididae) with extension of the noncoding region
Article information

Abstract
We present a nearly complete mitochondrial genome (mitogenome) of Hymenolepis diminuta from a Danish isolate, which was reassembled and comprehensively annotated using whole-genome sequencing data retrieved from the Sequence Read Archive (accession No. ERS056110). Although 2 mitogenomes of H. diminuta have previously been submitted to GenBank (accession No. AP017664, Danish isolate; NC_002767, putative German laboratory strain), the former lacks noncoding regions, while the latter harbors relatively short repeat units. Our newly reconstructed mitogenome is 14,090 bp long, comprising 12 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes, and a 643-bp noncoding region containing 7 tandem repeat units. The annotated sequence has been deposited in the Third Party Annotation database in GenBank (accession No. BK071817). Phylogenetic analysis based on mitogenomic sequences confirmed a close relationship between H. diminuta and Hymenolepis nana. This improved mitogenome sequence represents a valuable resource for comparative mitogenomic and phylogenetic investigations within the families Hymenolepididae, Taeniidae, and Diphyllobothriidae.
The rat tapeworm Hymenolepis diminuta (Cyclophyllidea: Hymenolepididae) is a widely studied model organism in experimental parasitology due to its noninvasive lifestyle, immunomodulatory properties, and tractable life cycle involving beetles and rodents as intermediate and definitive hosts, respectively [1]. Furthermore, it plays an important ecological role as a common parasite in wild rodent populations [2]. Beyond its biomedical relevance, H. diminuta has made significant contributions to understanding host–parasite interactions, gastrointestinal immune responses, and helminth-induced microbiome modulation. In particular, mitochondrial genome (mitogenome) data have provided valuable insights into the phylogenetics and evolution of cestodes. Two mitogenome sequences for H. diminuta were previously deposited in GenBank (AP017664, Danish isolate, 13,708 bp, Hd-p; NC_002767, putative German laboratory strain, 13,900 bp, Hd-c2 [3]). However, the Hd-p mitogenome lacks noncoding regions (NCRs), while the Hd-c2 mitogenome harbors relatively short repeat units (RUs). These NCRs, often housing tandem repeats and secondary structures, are becoming increasingly recognized as regulatory elements associated with mitochondrial replication and transcription. Given recent studies highlighting the functional importance of NCRs, extending mitogenome sequences using next-generation sequencing technologies has become increasingly important [4]. AT-rich repeats in NCRs not only regulate replication and transcription but also serve as valuable genetic markers for intraspecific variability and infection dynamics. This study aimed to reconstruct and analyze Hd-c1, a nearly complete mitogenome of H. diminuta from a Danish isolate, with a focus on extending and characterizing the previously unresolved NCR.
Whole-genome sequencing (WGS) data of H. diminuta were obtained from the 50 Helminth Genomes Project [5]. Illumina paired-end reads (2×100 bp) were downloaded from the Sequence Read Archive (accession No. ERS056110). Briefly, raw reads (37.11 Gb per paired-end sample) with a Phred quality score threshold of 33 that had been generated using an Illumina HiSeq 2000 platform were cleaned by adapter removal and quality trimming using Trimmomatic v0.39 [6]. The resulting high-quality reads were used to reconstruct a nearly complete mitogenome of H. diminuta. Among the genome assemblers evaluated in a benchmarking analysis [7], GetOrganelle v1.7.7.1 toolkit (https://github.com/Kinggerm/GetOrganelle) [8] demonstrated the best performance and was therefore selected for use in the genome assembly. Seed and label databases were customized using RefSeq mitogenomes of the class Cestoda (NCBI taxonomy ID 6199). The final assembly was visualized and manually validated in Bandage v0.8.1 [9] to assess any topological ambiguities.
Mitogenome annotation, including mitochondrial protein-coding genes (mPCGs) and functional RNAs, such as transfer RNA (tRNA) and ribosomal RNA, was performed using the MITOS2 pipeline through a GALAXY server v2.1.9 (https://usegalaxy.org/). The open reading frames of the mPCGs were manually verified using open reading frame finder from NCBI (https://www.ncbi.nlm.nih.gov/orffinder/), employing the invertebrate mitochondrial genetic code, and they were further identified by comparison with previously published mitogenomes of the family Hymenolepididae. tRNA genes undetected by MITOS2 were retrieved using BLASTn searches against the Rfam v15 database containing Hymenolepididae sequences [10]. RUs were identified using Tandem Repeats Finder v4.09.1 [11] with the default settings. The overall gene organization was visualized using Proksee v6.0.2 [12].
A total of 12 mPCGs, along with the individual nad1 and cox1 genes, were retrieved from GenBank for 11 platyhelminth species belonging to families Hymenolepididae, Taeniidae, and Diphyllobothriidae. The amino acid sequences were downloaded in FASTA format, and the mPCGs were concatenated to construct a combined dataset. Each dataset, including the concatenated mPCGs, nad1, and cox1, was aligned at the amino acid level using MAFFT v7 (https://mafft.cbrc.jp/) with the default settings. Phylogenetic trees were inferred using the maximum likelihood method and the Le_Gascuel_2008 model in MEGA11 [13]. Initial trees for the heuristic search were generated by applying the neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the Jones–Taylor–Thornton model and then selecting the topology with the superior log likelihood value. A discrete gamma distribution was used to model differences in evolutionary rates among sites (5 categories). Branch support was assessed by bootstrap analysis with 1,000 pseudoreplicates. Schistosoma mansoni and Schistosoma japonicum were designated as outgroups.
We used the GetOrganelle v1.7.7.1 toolkit [8], which performs preassembly filtering based on organelle-specific seed sequences, to facilitate mitogenome reconstruction from WGS data. The resulting well-annotated mitogenic circular sequence was 14,090 bp long (Fig. 1; Table 1). The assemblage was visually confirmed using Bandage v0.8.1 [9], with a sequencing depth ranging from 155.8-fold to 1,570.8-fold (Supplementary Fig. S1). The nearly complete annotated mitogenome sequence was deposited in the Third Party Annotation database of GenBank (accession No. BK071817).
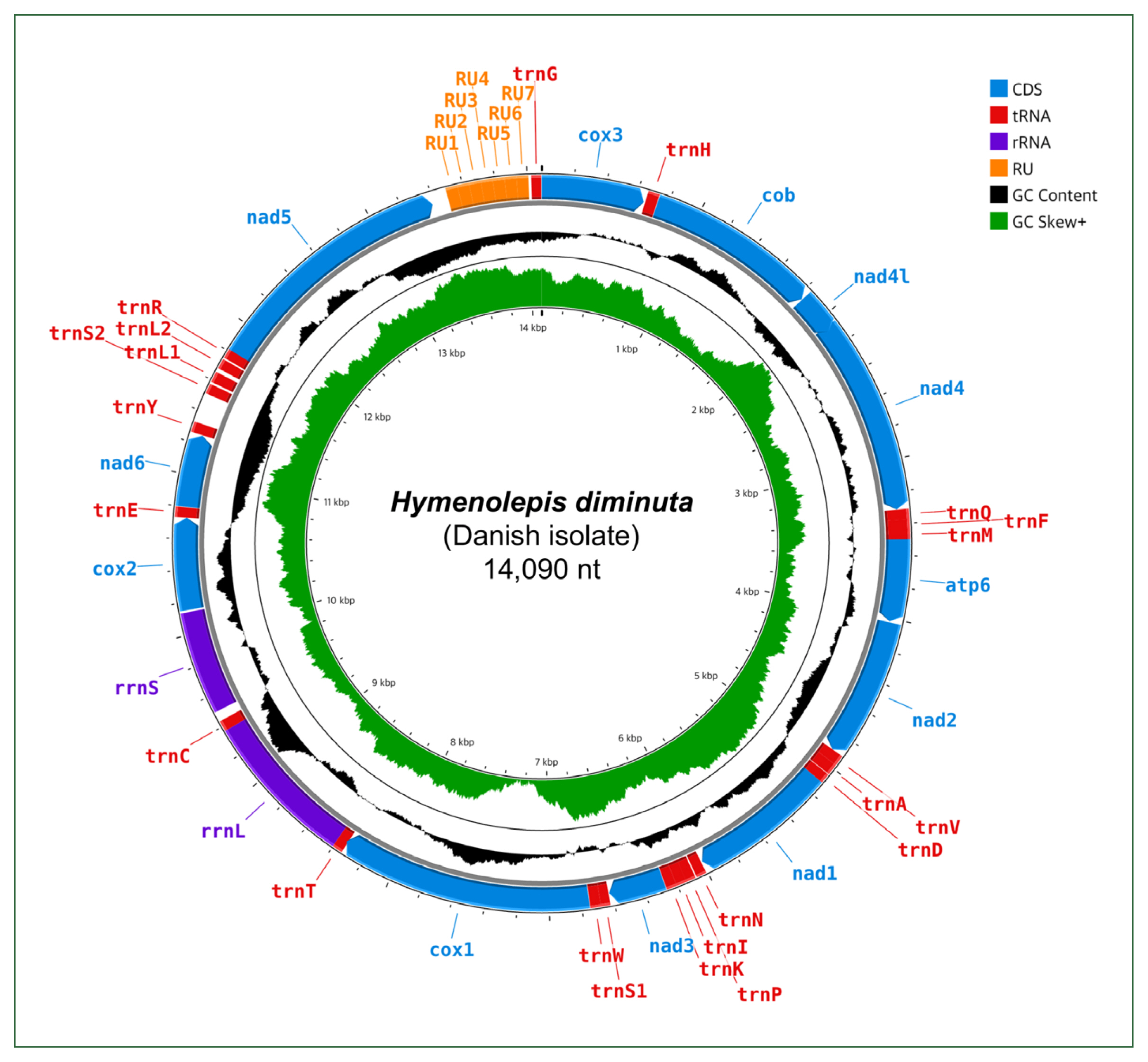
Annotated map of the nearly complete circular mitogenome of Hymenolepis diminuta (Danish isolate). CDS, coding sequence; tRNA, transfer RNA; rRNA, ribosomal RNA; RU, repeat unit; GC, guanine-cytosine.
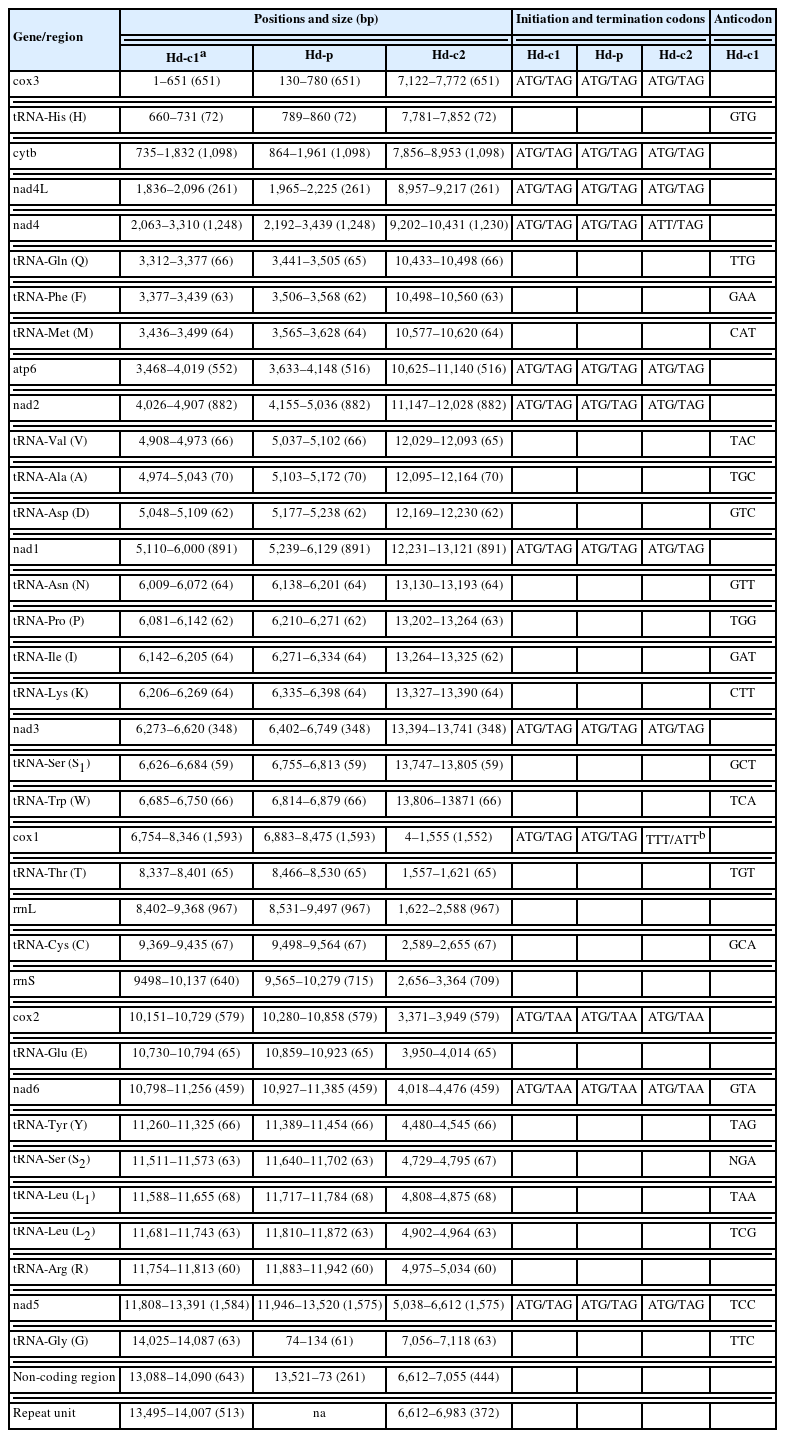
Gene content, length, initiation and stop codons of Hymenolepis diminuta mitogenomes from different datasets, including 2 complete mitogenomes (Hd-c1, BK071817 and Hd-c2, NC_002767) and 1 partial mitogenome (Hd-p, AP017664)
Pairwise alignment of our nearly complete mitogenome (Hd-c1) with the previously deposited complete (Hd-c2) and partial (Hd-p) mitogenomes of H. diminuta revealed highly conserved genomic content with minimal gene-level divergence. However, the 3 mitogenomes did not show any sequence similarities in the NCR, as confirmed by BLASTn (Supplementary Fig. S2). The lack of similarity between Hd-c1 and Hd-p (both of Danish origin) is attributable to the absence of RUs and a truncated NCR in Hd-p. In contrast, the divergence between Hd-c1 (Danish isolate) and Hd-c2 (putative German laboratory isolate) results from differences in both NCR length and RU composition. Accordingly, the improved nearly complete mitogenome (Hd-c1) reported in this study provides a reliable and valuable resource for future mitogenomic research in the family Hymenolepididae.
Consistent with the mitogenomes of the other H. diminuta strains, our nearly complete H. diminuta mitogenome (Hd-c1) included 12 mPCGs (nad1–6, nad4L, cox1–3, cytb, and atp6), 2 ribosomal RNA genes (rrnL and rrnS), and 22 tRNA genes (Table 1). Notably, Hd-c1 included a 643-bp NCR housing 7 RUs (each 73 bp) (Supplementary Fig. S3). The total length of the mPCGs was 10,100 bp, accounting for 71.68% of the nearly complete mitogenome. cox1 (1,593 bp) was the longest and nad4L (261 bp) was the shortest mPCG. ATG and TAG were the most common codons for initiation and termination, respectively. In all 3 mitogenomes, TAA was only used as a termination codon in cox2 and nad6, while TTT/ATT were observed exclusively as noncanonical codons in Hd-c2. The total length of the tRNA genes was 1,422 bp, ranging from 59 nucleotides (tRNA-Ser1) to 72 nucleotides (tRNA-His) in the nearly complete mitogenome (Hd-c1). Hd-c1 had an overall gene length pattern similar to Hd-p, with slight differences in atp6 (516 bp), rrnS (715 bp), and nad5 (1,575 bp). Compared with Hd-c2, Hd-c1 showed slight differences with nad4 (1,230 bp), atp6 (516 bp), cox1 (1,552 bp), rrnS (709 bp), and nad5 (1,575 bp). The NCR (643 bp) of the nearly complete mitogenome (Hd-c1) contained a 513-bp segment comprising 7 73-bp RUs, whereas Hd-p included a shorter NCR (261 bp) lacking any RUs (Table 1; Supplementary Fig. S3). In contrast, Hd-c2 harbored a 444-bp NCR that housed a relatively short 372-bp segment comprising 12 31-bp RUs [3]. The putative roles of the NCR suggest that highly repetitive AT-rich regions may influence mitochondrial function by acting as initiation or termination sites for DNA replication and transcription [4]. Such structural and functional variability may potentially be linked to longevity, underscoring the need for more comprehensive investigations into the biological significance of NCRs in parasitic helminths [14].
The genetic differences of 12 mPCGs in the mitogenomes of 11 representative species of the subclass Eucestoda were compared at the nucleotide level (Table 2). Although the lengths of nad4L and nad6 were identical between Hd-c1 and Hd-c2, they showed a nucleotide sequence divergence of 0.77% and 0.66%, respectively. The sequence differences between the nearly complete mitogenome (Hd-c1) of H. diminuta and one Hymenolepididae species, 5 Taeniidae species, and 3 Diphyllobothriidae species ranged from 16.44% (Hymenolepis nana) to 57.90% (Spirometa erinaceieuropaei). Substantial sequence differences were detected in nad6 (57.90%), cox3 (55.45%), nad5 (53.51%), and apt6 (51.92%).
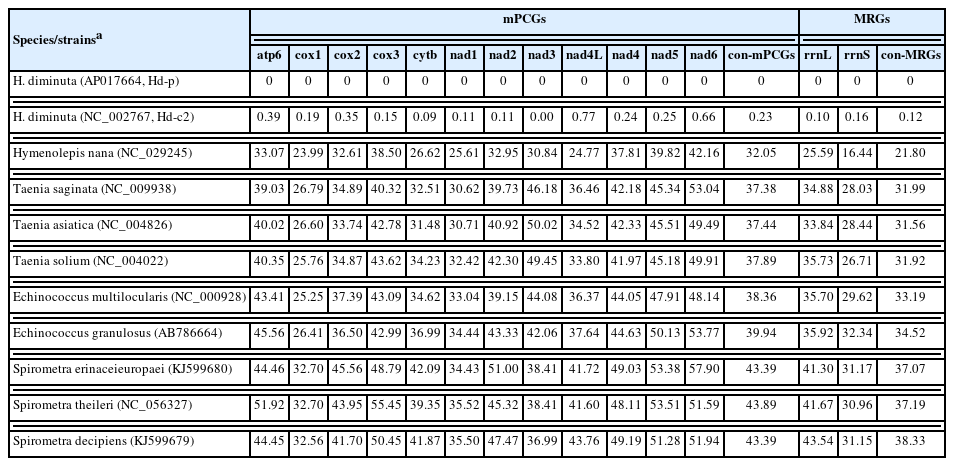
Estimation of pairwise distances rate (%) of individual and concatenated mPCGs and MRGs between Hymenolepis diminuta and members of the families such as Hymenolepididae, Taeniidae, and Diphyllobothriidae
The phylogeny among Hymenolepis species was well-resolved, with strong nodal support (Fig. 2). The H. diminuta mitogenomes (Hd-c1, Hd-p, and Hd-c2) clustered together as a distinct clade, showing a close relationship with H. nana. Similar phylogenetic topologies were consistently recovered across trees constructed from the 12 concatenated mPCGs, as well as from individual cox1 and nad1 sequences.
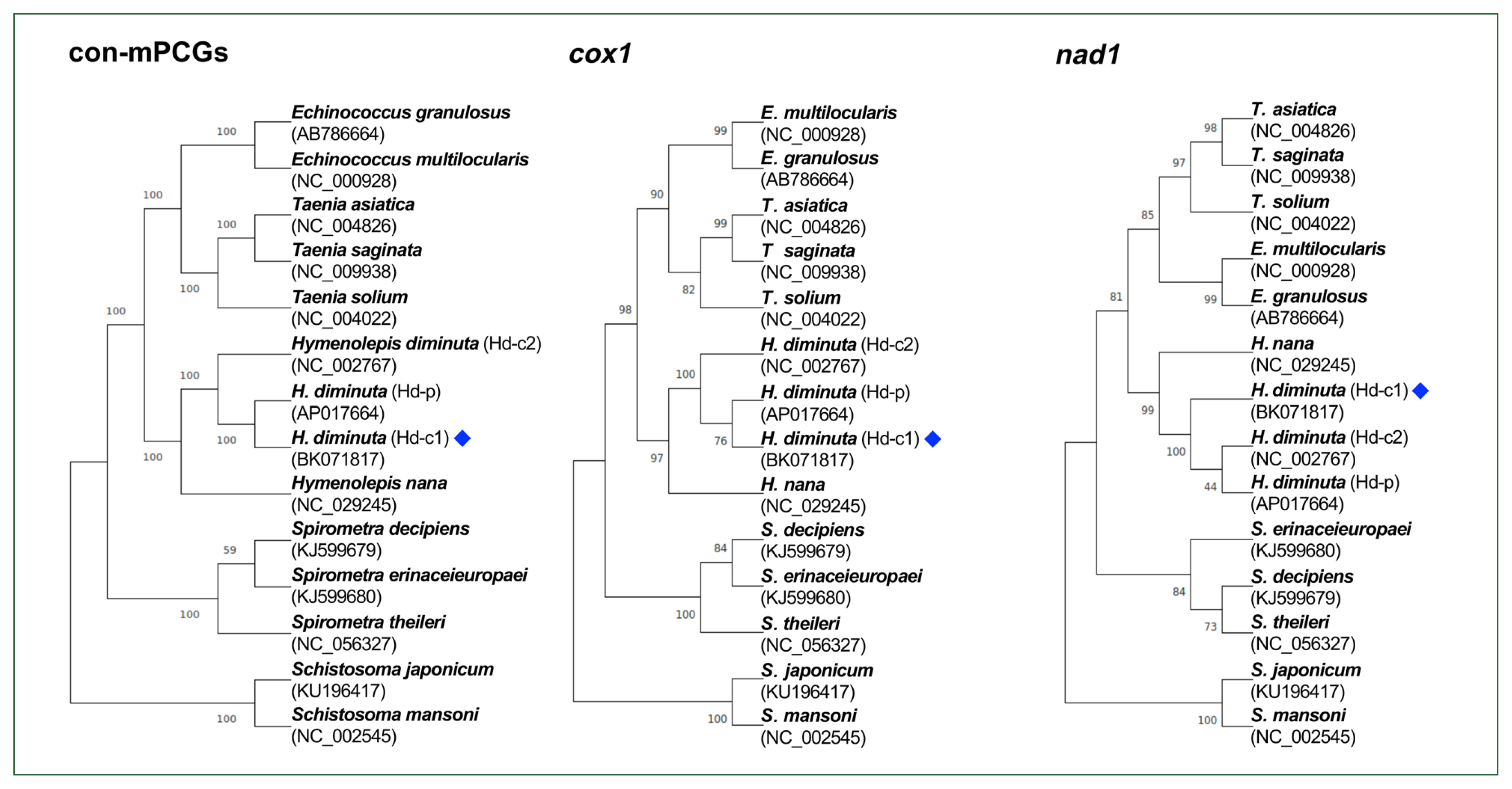
Phylogenetic tree showing the position of Hymenolepis diminuta. The mitogenome generated in this study is indicated by a blue diamond. The tree is based on amino acid sequences derived from the concatenation of 12 mitochondrial protein-coding genes (mPCGs), cox1, and nad1. Schistosoma mansoni and Schistosoma japonicum were used as outgroups. Nodal support values, estimated from 1,000 bootstrap pseudoreplicates, are indicated at each node. GenBank accession numbers are listed alongside the corresponding species names.
To summarize, we present Hd-c1, an improved and nearly complete mitogenome of H. diminuta, which includes a fully resolved NCR containing 7 RUs. This represents a substantial improvement over previously deposited sequences (AP017664 and NC_002767), in which the RU was either absent or only comprised short segments. Our study underscores the value of recently developed mitochondria-specific assembly pipelines, which are expected to facilitate the generation of high-quality mitogenomes from cestode-derived WGS datasets. This newly assembled H. diminuta mitogenome is a reliable resource for future comparative mitogenomic and phylomitogenomic investigations within Hymenolepididae and related cestode families.
Notes
Author contributions
Conceptualization: Cho K, Choi YK, Yoo WG
Data curation: Cho K
Funding acquisition: Choi YK, Yoo WG
Investigation: Cho K
Methodology: Cho K, Kim M
Project administration: Choi YK, Yoo WG
Software: Cho K
Supervision: Yoo WG
Validation: Kim M
Visualization: Cho K
Writing – original draft: Cho K, Yoo WG
Writing – review & editing: Kim M, Choi YK, Yoo WG
Conflict of interest
The authors declare no conflict of interest related to this study.
Acknowledgments
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (Ministry of Science and ICT) (grant No. RS-2022-NR070067 and RS-2024-00509361) (http://www.nrf.re.kr) and was partially supported by the Research Institute for Veterinary Science, Seoul National University.
Supplementary Information
Supplementary material is available with this article at https://doi.org/10.3347/PHD.25061.