Genome-wide identification of histone lysine methyltransferases and their implications in the epigenetic regulation of eggshell formation-related genes in a trematode parasite Clonorchis sinensis
Article information

Abstract
Epigenetic writers including DNA and histone lysine methyltransferases (DNMT and HKMT, respectively) play an initiative role in the differentiation and development of eukaryotic organisms through the spatiotemporal regulation of functional gene expressions. However, the epigenetic mechanisms have long been suspected in helminth parasites lacking the major DNA methyltransferases DNMT1 and DNMT3a/3b. Very little information on the evolutionary status of the epigenetic tools and their role in regulating chromosomal genes is currently available in the parasitic trematodes. We previously suggested the probable role of a DNMT2-like protein (CsDNMT2) as a genuine epigenetic writer in a trematode parasite Clonorchis sinensis. Here, we analyzed the phylogeny of HKMT subfamily members in the liver fluke and other platyhelminth species. The platyhelminth genomes examined conserved genes for the most of SET domain-containing HKMT and Disruptor of Telomeric Silencing 1 subfamilies, while some genes were expanded specifically in certain platyhelminth genomes. Related to the high gene dosages for HKMT activities covering differential but somewhat overlapping substrate specificities, variously methylated histones were recognized throughout the tissues/organs of C. sinensis adults. The temporal expressions of genes involved in eggshell formation were gradually decreased to their lowest levels proportionally to aging, whereas those of some epigenetic tool genes were re-boosted in the later adult stages of the parasite. Furthermore, these expression levels were significantly affected by treatment with DNMT and HKMT inhibitors. Our data strongly suggest that methylated histones are potent epigenetic markers that modulate the spatiotemporal expressions of C. sinensis genes, especially those involved in sexual reproduction.
Introduction
Chromosomal DNAs in eukaryotic cells are present in a highly organized structure by being packaged into nucleosomes, the structural unit of chromatin, in combination with histone proteins [1]. Together with the histone globular domains, the unstructured side chains of specific amino acid (aa) residues provide binding platforms for various chromatin-binding proteins including chromatin remodelers, DNA/histone-modifying enzymes, and general transcription factors [2]. The physicochemical nature of globular domains and side chains profoundly affect the macroscopic chromatin structure and gene expression by modulating attachments of structural/functional proteins involved in the biological processes. The nucleosomal histones are modified by adding a wide variety of chemical groups such as methyl, acetyl, phosphoryl, propionyl, serotonyl, and acyl groups, to meet the essential biological demands [2,3]. Mis-regulated histone modifications may result in inappropriate activation or repression of various genes, which can lead to developmental anomalies and/or pathobiological progression of various diseases including cancer [4].
Epigenetics refers to the heritable modification of gene expression without any change in DNA sequences, thereby introducing diverse phenotypes among cells with identical genetic backgrounds in multicellular organisms. The chromosomal events are also implicated in maintaining the integrity and stability of gene expression profiles in the resulting variants [5,6]. During the epigenetic processes, gene expression is regulated at various levels in response to the DNA and histone modifications, as well as the actions of non-coding micro RNAs [5]. Of the diverse histone modifications, methylation and acetylation are the most well studied. Reversible addition or removal of methyl and acetyl groups induces a structural shift in chromatin from compact fibers to relaxed ones, or vice versa. These conformational alterations are closely related to the modulation of the frequency, activity, or extent of nearby gene expressions by influencing the accessibility of the transcriptional machinery [2].
A series of methylated Lys residues associated with the epigenetic mechanisms have been reported in nucleosomal histones, such as mono (me1), di (me2), and tri (me3)-methylation of Lys residues at positions 4, 9, 27, 36, and 79 of histone H3 (H3K4, H3K9, H3K27, H3K36, and H3K79); at positions 20 and 59 of histone H4 (H4K20 and H4K59); and at position 26 of histone H1B [2,7]. The methyl groups are transferred from S-adenosyl methionine (SAM) to the ε-amino group of Lys, and the biochemical reactions are catalyzed by a group of enzymes, collectively termed histone Lys methyltransferase (HKMT) [8]. Except for the Disruptor of Telomeric Silencing1 (DOT1) family, which is responsible for the methylation of H3K79, almost all HKMT proteins possess a relatively conserved domain, the suppressor of variegation (Su(var)3–9), enhancer of zeste (E(z)), and trithorax (SET) [7]. The methylated histones may specify both active and repressed chromatin states, depending on the positions of modified Lys residues and the degree of methylation. However, the biological role of each HKMT remains elusive [2,7]. In general, H3K4me3, H3K79me2, and H4K20me1 are involved in transcriptional activation, whereas H3K9me2, H3K9me3, and H3K27me3 are associated with gene silencing [2,9].
Protein genes involved in epigenetic processes, such as those encoding DNA methyltransferase (DNMT) and HKMT, show punctate distribution patterns across the genomes of eukaryotic taxa [10,11]. This suggests that eukaryotic organisms have uniquely evolved epigenetic toolkits to meet individual requirements for adapting to different environmental conditions and/or regulating germline/soma differentiation. Defense against mobile genetic elements provides an additional driving force for the mosaic evolutionary pathway. The presence and biological implications of the epigenetic processes, or at least methylated cytosine-mediated gene silencing mechanism, have been highly suspected in lower animals, including those of the phylum Platyhelminthes, owing to the lack of DNMT1 (maintenance of cytosine methylome) and DNMT3 (de novo generation of cytosine methylome)-like enzymes [10–12]. However, recent studies have shown that biochemical modifications of DNA and histones are involved in the spatiotemporal regulation of gene expression without genetic alterations in platyhelminth species [13,14].
Clonorchis sinensis belonging to the class Trematoda of the phylum Platyhelminthes parasitizes the biliary tracts and causes clonorchiasis in humans. Clonorchiasis poses a great socio-economic burden in endemic areas, including China, Korea, and Vietnam [15]. Humans are accidentally affected by the parasitic disease by consuming raw or undercooked freshwater fish containing C. sinensis metacercariae. The metacercariae excyst in the duodenum and grow into mature adults in the bile ducts. Patients with clonorchiasis exhibit a wide spectrum of clinical symptoms, ranging from obstructive cholangiopathy to cholangiocarcinoma, depending on the rate and duration of infection [16]. The hepatobiliary worm is categorized as Group I biocarcinogens by the World Health Organization [17]. Similar to other digenean parasites, C. sinensis has a complex life cycle and undergoes metamorphosis in its respective hosts under different biophysicochemical environments: 4 stages from miracidia to cercaria in snail, the metacercaria stage in freshwater fish, and the juvenile/adult stage in mammal [18]. Furthermore, approximately 32.0% of the parasite genome comprises diverse repetitive elements, including long terminal repeat (LTR) and non-LTR retrotransposons [19,20]. In addition to the parasitic life mode, this genomic feature implies that certain types of molecular mechanisms operate effectively in C. sinensis for regulating gene expression in a timely manner and/or defensing against the selfish intragenomic invaders [10]. Reversible modification of the chromatin structure induced by methylated DNA and/or histones may be a strong candidate for the epigenetic gene regulation. We have previously reported the structural and biochemical properties of a DNMT homolog in C. sinensis [21]. In this study, we analyzed HKMT proteins by screening the entire C. sinensis genome. Temporal expression patterns of these HKMT genes and the effects of methyltransferase inhibitors on the expression of several functional genes were also investigated in the liver fluke to determine their role as epigenetic regulators.
Materials and Methods
In silico identification of SET-HKMT proteins
The genomic and proteomic databases of C. sinensis (Platyhelminthes: Trematoda: Opisthorchiida) in GenBank (https://www.ncbi.nlm.nih.gov/genbank/) were searched using the BLAST programs with the aa sequences of human SET-HKMT and DOT1 family members [7,22]. The sequences of C. sinensis proteins retrieved by the similarity searches were used as queries in a second series of BLAST tests against the C. sinensis databases. Homologous sequences were similarly isolated from the genomic databases of Schmidtea mediterranea (Platyhelminthes: Turbellaria: Tricladida) in the GenBank and Schistosoma mansoni (Platyhelminthes: Trematode: Strigeidida), Hymenolepis microstoma (Platyhelminthes: Cestoda: Hymenolepididae), Echinococcus granulosus (Platyhelminthes: Cestoda: Taeniidae), and Globodera pallida (Ecdysozoa: Nematoda: Rhabditida) in the GeneDB (https://www.genedb.org/) (E-value cutoff, 1×10−5). Functional domains including SET were annotated by analyzing the retrieved sequences using the Simple Modular Architecture Research Tool (SMART, http://smart.embl-heidelberg.de). The schematic diagrams of the preserved domains of each protein were constructed using the SMART program.
Phylogenetic analysis
The aa sequences of the SET domains from a total of 126 proteins were aligned and manually trimmed using the ClustalX (ver. 2.1) and GeneDoc programs, respectively. The alignment was applied in the construction of a maximum likelihood tree using the PhyML (ver. 3.1) [23] or MEGA X program [24]. The analytical parameters were as follows: Jones-Taylor-Thornton model for aa substitution, gamma distribution rates with invariant sites (4 gamma categories), and partial deletion of gaps/missing data (site coverage cutoff, 95.0%). The gamma rate and proportion of invariant sites were empirically estimated using the initial alignment. The statistical significance of internal branching nodes was evaluated using the non-parametric Shimodaira-Hasegawa-like approximate likelihood ratio test (SH-aLRT) or the standard bootstrapping test of 1,000 replicas. The resulting trees were displayed using the TreeView program.
Immunological detection of methylated histone derivatives
C. sinensis adults (42-day-old) were collected from experimentally infected rats, as described previously [25]. The worms (>30 worms) were minced with a razor blade and homogenized in Triton extraction buffer (TEB: PBS containing 0.5% (v/v) Triton X-100 and a protease inhibitor cocktail (complete; Roche Diagnostics GmbH, Mannheim, Germany)) using a Dounce tissue grinder with a loose pestle (Type A; Wheaton, Millville, NJ, USA). The homogenate was centrifuged at 10,000 g for 20 min at 4°C, and the supernatant was stored as the cytosolic protein extract. After washing with TEB, the pellet enriched with nuclei was resuspended in 0.2 N HCl and incubated overnight at 4°C. The solution was centrifuged at 20,000 g for 5 min at 4°C, resulting in the nuclear protein extract. The cytosolic and nuclear protein extracts were similarly prepared from HeLa cells and the liver tissue of a normal rat. Total protein in the protein extracts was quantified using the Pierce BCA assay kit (Thermo Fisher Scientific, Rockford, IL, USA). All the protein samples (10 μg) were electrophoresed on 15.0% SDS-PAGE gels under reducing conditions and transferred onto nitrocellulose membranes (Schleicher & Schuell Bioscience, Dassel, Germany). The membranes were reacted with antibodies specific to histone H3 or its methylated derivatives (Supplementary Table S1) and then, with horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse IgG antibodies (Bethyl Laboratories, Montgomery, TX, USA). Positive signals were visualized using an enhanced chemiluminescence detection system (GE Healthcare, Pittsburgh, PA, USA). Images were captured using a LAS-400 Lumino image analyzer (Fujifilm, Tokyo, Japan).
The whole bodies of C. sinensis adults were fixed in paraffin blocks and sliced onto electrostatically charged glass slides (Superfrost Plus, Manzel-Glaser, Germany). After deparaffinization and rehydration according to the manufacturer’s instructions (DAKO, Carpinteria, CA, USA), the slides were soaked in a 3.0% H2O2 solution for 5 min and then in TBS/T buffer with 3.0% BSA for 1 h. The sections were incubated overnight at 4°C in the BSA-TBS/T buffer with each of the antibodies listed in Supplementary Table S1 (1:200 dilution). The primary antibodies bound to the worm sections were detected using HRP-conjugated secondary antibodies (1:1,000 dilution; Cappel, West Chester, PA, USA) and the HIGHDEF blue chromogen substrate (Enzo Life Sciences, Farmingdale, NY, USA). A mixture of pre-immune rabbit serum (n=3, 1:200 dilution) was used as a negative control for the immunohistochemical staining.
Temporal expression profiles of C. sinensis genes
Total RNAs were extracted from C. sinensis worms at various stages (>30 worms/stage) using QIAzol solution and the RNeasy Mini kit (Qiagen, Hilden, Germany). After removing residual genomic DNAs using RNase-free DNase (New England Biolabs, Ipswich, MA), Complementary DNAs (cDNAs) were synthesized from the RNAs using the iScript cDNA synthesis kit (Bio-Rad, Munich, Germany). The numbers of epigenetic tool gene and reproduction-related gene transcripts were examined via quantitative real-time PCR (RT-qPCR) using the cDNA samples and gene-specific primers (Supplementary Table S2). qPCR was conducted using the SYBR Green Master Mix and the CFX96 detection system according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA, USA). The presence of a single-target amplicon was confirmed by analyzing melting curves. Each reaction was conducted in triplicate, and the relative transcription levels against a reference β-actin gene (DF143505) were presented as the mean±SD (ΔCq calculation method) [26]. A Student’s t test was used for statistical analyses, and a probability level of P<0.05 was considered statistically significant.
Methyltransferase inhibitors and gene expressions
Live C. sinensis worms (42- and 140-day-old, 30 worms/group) were incubated in RPMI-1640 media (phenol red- and serum-free, pH 7.2) supplemented with 5 μm SGI-1027 (Selleck, Houston, TX, USA) or 0.5 μm 3-deazaneplanocin A (DZNep; Selleck) at 37°C in a 5.0%-CO2 incubator, by refreshing the media daily. After a 5-day incubation, the total RNAs were extracted from the worms for gene expression analysis using RT-qPCR, as described above. The responsive gene expression levels in the experimental groups were calculated as fold changes against those in a control group, which was not treated with the chemicals (ΔΔCq calculation method) [26].
Results
Identification of SET-HKMTs in C. sinensis and other helminths
Based on previous reports [7,22], several human SET-HKMTs were selected to represent each of the diverse HKMT families. The aa sequences of these human proteins were used as queries in BLAST searches against the C. sinensis proteomic database in GenBank (BioProject No. PRJNA563175). The GenBank database was further surveyed for isolating Clonorchis proteins named HKMT or HMT by the original authors. The retrieved aa sequences were compared to one another to remove redundant entries (cutoff, 95.0%), and a total of 32 proteins were finally identified as the non-allelic HKMT paralogs of C. sinensis (Table 1). Information on orthologous proteins was similarly obtained from other trematode (24 proteins in S. mansoni), cestode (28 proteins in E. granulosus and 41 proteins in H. microstoma), turbellarian (7 proteins in S. mediterranea), and nematode (20 proteins in G. pallida) species, for which genome sequences had been drafted, using the human and C. sinensis SET-HKMT sequences as queries (Supplementary Tables S3-S7).
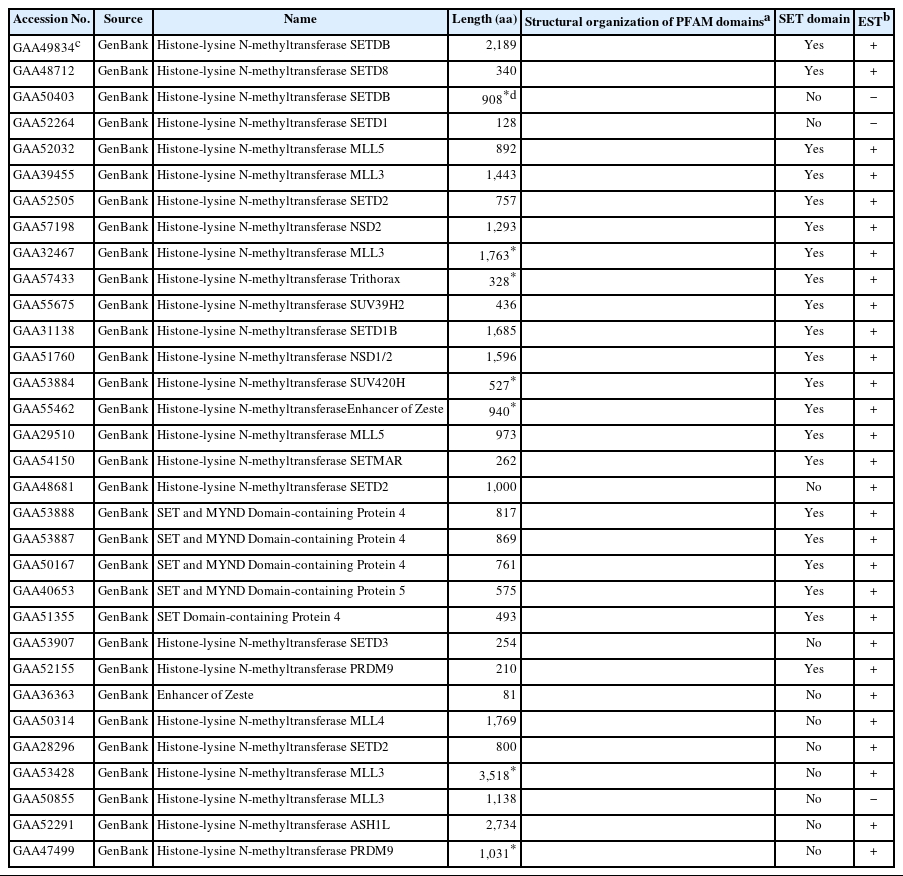
SET-HKMT family proteins idnetified in the proteomic database of Clonorchis sinensis
Structural characters of the helminth SET-HKMTs
The Clonorchis SET-HKMT proteins were assigned to multiple SET-HKMT families, including SUV39, SET1, SET2, and SMYD (Table 1). Screening of the C. sinensis transcriptomic database in GenBank with the SET-HKMT sequences demonstrated that the majority of C. sinensis proteins, except for GAA50403, GAA52264, and GAA50855, are expressed at least at the mRNA level. The SMART program recognized aa stretches preserved in various functional domains such as SET, plant homeodomain (PHD), and Set2-Rpb1 interaction (SRI) in their primary structures, although the main SET domain could not be defined in 11 sequences (Table 1). The highly polymorphic lengths (81–3,518 residues) and lack of the SET domain indicated that at least some of protein sequences had been incompletely predicted.
The Clonorchis SET domain sequences were aligned with one another, and their aa conservation patterns were examined by referring to previous reports [7,27]. The SET domain sequences were highly diverged, and the similarity values ranged from 2.0% (GAA49834 vs GAA52155) to 67.1% (GAA32467 vs GAA39455) (average±SD, 18.2%±0.10). Short conserved aa stretches, which are involved in the binding of SAM and/or histone Lys [7,27], were readily detected in the SET-N and SET-C domains of these C. sinensis proteins (Motifs I – IV in Fig. 1). However, critical amino acids involved in the formation of H-bonds/van der Waals interactions with the SAM molecule (Gly, Asn, and His indicated by open arrowheads) or the ε-amino group of histone Lys (Tyr indicated by solid arrowheads) were substituted by other amino acids in some of the C. sinensis proteins. Similar aa conservation patterns were observed in the SET domain sequences identified in other helminths (data not shown).
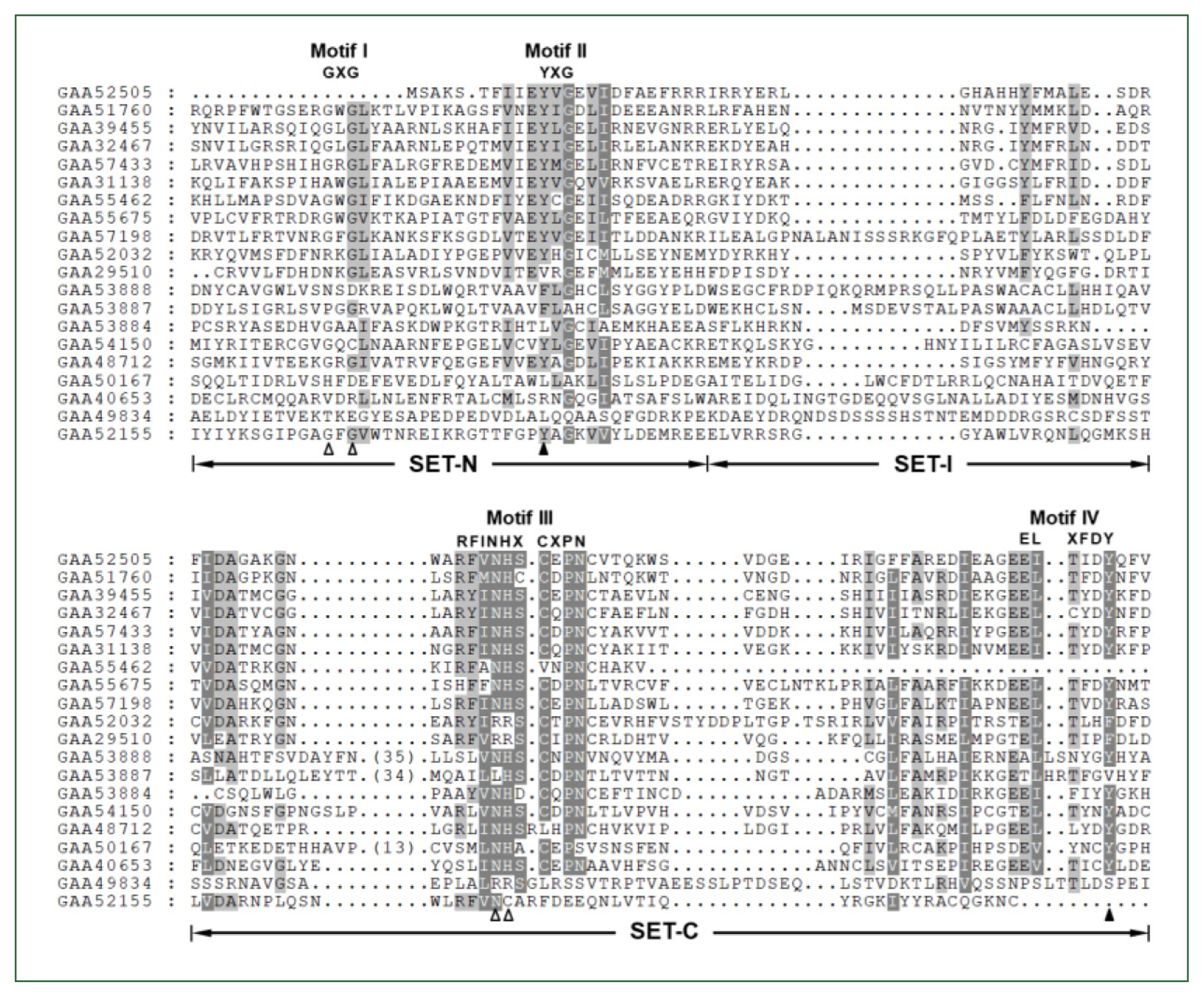
Comparison of SET domain sequences conserved in the Clonorchis sinensis histone lysine methyltransferase (CsHKMT) proteins. The amino acid (aa) sequences of CsHKMT SET domains, which were defined in the full CsHKMT sequences using the SMART program, were aligned against one another for comparison of their primary structures. The degrees of the aa conservations are highlighted in different shades of grey, and gaps represented by dots were introduced in the alignment to increase the similarity values. The short aa signatures of the SET domain involved in the binding of S-adenosyl methionine (SAM) and histone Lys are presented above the alignment (Motifs I–IV). The solid triangle indicates the invariant Tyr forming H-bond/van der Waals interaction with the ε-amino group of histone Lys. Open arrowheads represent the positions of Gly, Asn, and His participating in the formation of the non-covalent bonds with SAM molecule.
Phylogeny of the helminth SET-HKMTs
The genomes of the helminth species encoded diverse SET-HKMTs, although some entries were not completely predicted (entries marked with asterisks in Table 1). The evolutionary relationships of these SET-HKMT orthologs were examined via phylogenetic analysis based on their SET domain sequences (21, 16, 18, 28, 4, 14, and 25 proteins from C. sinensis, S. mansoni, E. granulosus, H. microstoma, S. mediterranea, G. pallida, and human, respectively). As shown in Fig. 2, helminth proteins were clustered into tight clades together with human proteins, consistent with their respective names, whereas some clades, such as NSD2, SMYD4, SMYD5, and SETD4 comprised only helminth proteins. In addition, the genes encoding EHMT1 and SMYD4 had been specifically multiplied in G. pallida (solid arrowhead in Fig. 2) and H. microstoma (open arrowhead), respectively. Considering the fact that a considerable number of SET-HKMTs lacking the SET domain were excluded in the phylogenetic analysis, the major SET-HKMT clades were likely to have diverged early in bilaterian evolution, at least prior to the divergence of deuterostomes and protostomes.
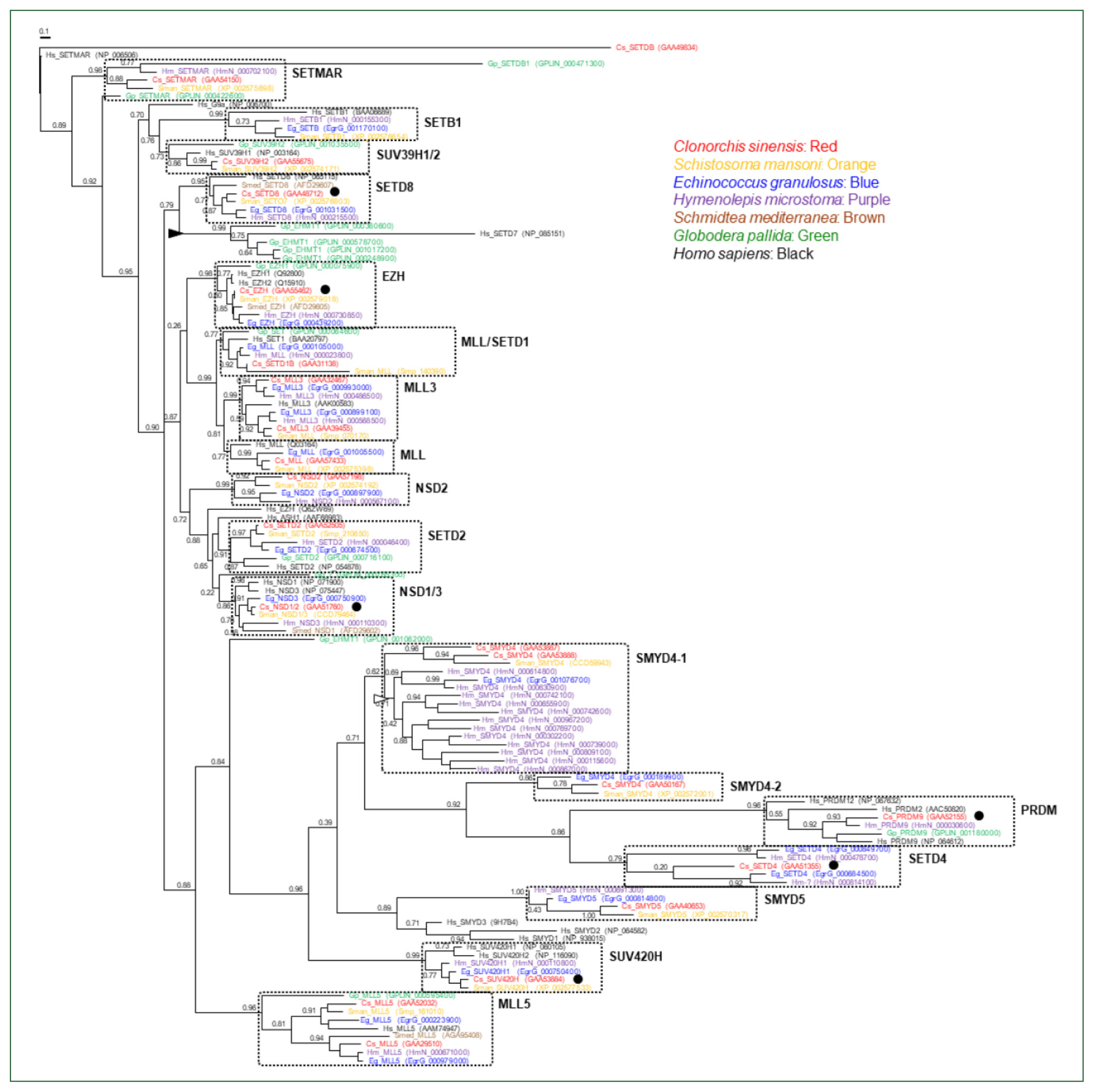
Phylogeny of Clonorchis sinensis histone lysine methyltransferases (HKMTs) and their platyhelminth and nematode orthologs. The amino acid sequences of HKMT SET domains identified in trematodes (C. sinensis [red] and Schistosoma mansoni [orange]), cestodes (Echinococcus granulosus [blue] and Hymenolepis microstoma [purple]), turbellarian (Schmidtea mediterranea [brown]), nematode (Globodera pallida [green]), and human (black) were aligned and used in the phylogenetic analysis. The unrooted maximum likelihood tree was constructed using PhyML. Branch support values obtained using the Shimodaira-Hasegawa-like approximate likelihood ratio test are indicated at the corresponding branching nodes. Proteins, coding genes of which appeared to be expanded specifically in G. pallida and H. microstoma, are indicate with solid and open arrowheads, respectively. The solid circles indicate C. sinensis proteins selected for the gene expression analyses.
Retrieving DOT1 protein sequences from the helminth proteomes
BLASTp searches using the aa sequence of human DOT1 (1,537 aa; GenBank accession number NP_115871.1) retrieved a single non-redundant protein from each of the trematode and cestode proteomic databases in the GenBank (E-value <2×10−11). Similar to SET-HKMTs, the lengths of these helminth proteins also varied ranging from 218 (Schistocephalus solidus DOT1; VDL97768.1) to 1,756 aa (E. granulosus DOT1; XP_024351501.1), the majority of which were likely to be partial. In the C. sinensis database, 2 proteins with 1,243 (RJW63735.1) and 1,704 (GAA52952.1) overlapping aa sequences significantly matched to the human DOT1 protein (E-values, 1×10−109 and 3×10−105, respectively). No homologous protein was retrieved from the current S. mediterranea proteome. A similarity search of the entire genomic sequences of the turbellaria using tBLASTn algorithm isolated several short DNA segments, of which theoretically translated proteins were matched to the human protein (query coverage <14.0%). Given the low coverage and/or the locations of matched sequences in the 3′-terminus of respective contigs as well as the low SET-HKMT protein number, the retrieval failure occurred probably due to the incomplete assembly of the flat worm genome.
Comparative analysis of the helminth DOT1s
Although partial, the catalytic DOT1 domain (PFAM id. PF08123) was readily recognized in the primary structures of these helminth DOT1 homologs (Fig. 3A). Some of these DOT1 motif sequences were aligned to that of a previously characterized human DOT1 protein [28]. As shown in Fig. 3B, the full catalytic core sequences were relatively well-preserved among them and exhibited identity values ranging from 36.0% (EgDOT1 vs. BxDOT1) to 100.0% (CsDOT1 vs OvDOT1) (average±standard deviation, 62.9±0.14%). The aa conservation was much more prominent in the segmental motifs constituting the SAM-binding pocket (Motifs I, I′, II, D1, and D2). The aa residues implicated in either the direct H-bond interaction with the cofactor SAM (solid circles in Fig. 3B) or the constitution of an access channel for H3K79 (open circles) were tightly conserved between the helminth and human orthologs. Identical amino acids may be involved in the formation of a channel leading to the inside of the SAM-binding pocket (solid arrowheads) and the regulation of the Lys-binding channel gate (open arrowheads), as described in the human DOT1 protein [28]. In addition to the DOT1 domain, several SCOP domains, including d1quua1 (2 central spectrin-like repeats of the σ-actin family), d1cuna2 (repeats of the σ-spectrin family), d1fcha (a domain found in the peroxisomal targeting signal 1 receptor), and d1eq1a (a domain found in the apolipophorin-III family), were occasionally detected in the primary structures of some DOT1 homologs. The human DOT1 protein further preserves the SMART AT_hook domain (id. SM00384), which is a DNA-binding motif with a preference for A/T-rich regions (Fig. 3A).

Structural characterization of Clonorchis sinensis DOT1 (CsDOT1) proteins. (A) The functional domains conserved in CsDOT1 were predicted using the SMART program and compared with those of its orthologs. The domain legends are shown at the bottom. (B) The SET domain sequences of DOT1 homologs shown in panel A were aligned and the degrees of amino acid (aa) conservation were differentially highlighted in the alignment. The secondary structure of the SET domain, which has been determined in human DOT1, is presented at the bottom of each alignment (rod, α-helix; arrow, β-sheet). The segmental motifs constituting the S-adenosyl methionine (SAM)-binding pocket (Motifs I, I′, II, D1, and D2) are shown on the top. Solid and open circles represent aa residues involved in the binding of SAM and Lys79 of histone H3, respectively. Amino acids involved in the formation of a channel leading to the inside of the SAM binding pocket and the regulation of the Lys-binding channel gate are denoted with solid and open arrowheads, respectively. The numerals in the alignment are the positions of corresponding residues in human DOT1 (NP_115871.1).
The aa sequences of platyhelminth DOT1 proteins were aligned with those of nematode and deuterostomian orthologs, as well as cnidarian proteins (Nematostella venctensis and Hydra vulgaris DOT1), which were selected to comprise an outgroup. Using the sequence alignment, a maximum likelihood tree was constructed to examine their evolutionary relationships. In the tree rooted with the cnidarian proteins, DOT1 homologs showed a clustering pattern corresponding to the phylogeny of their donor organisms (Fig. 4).
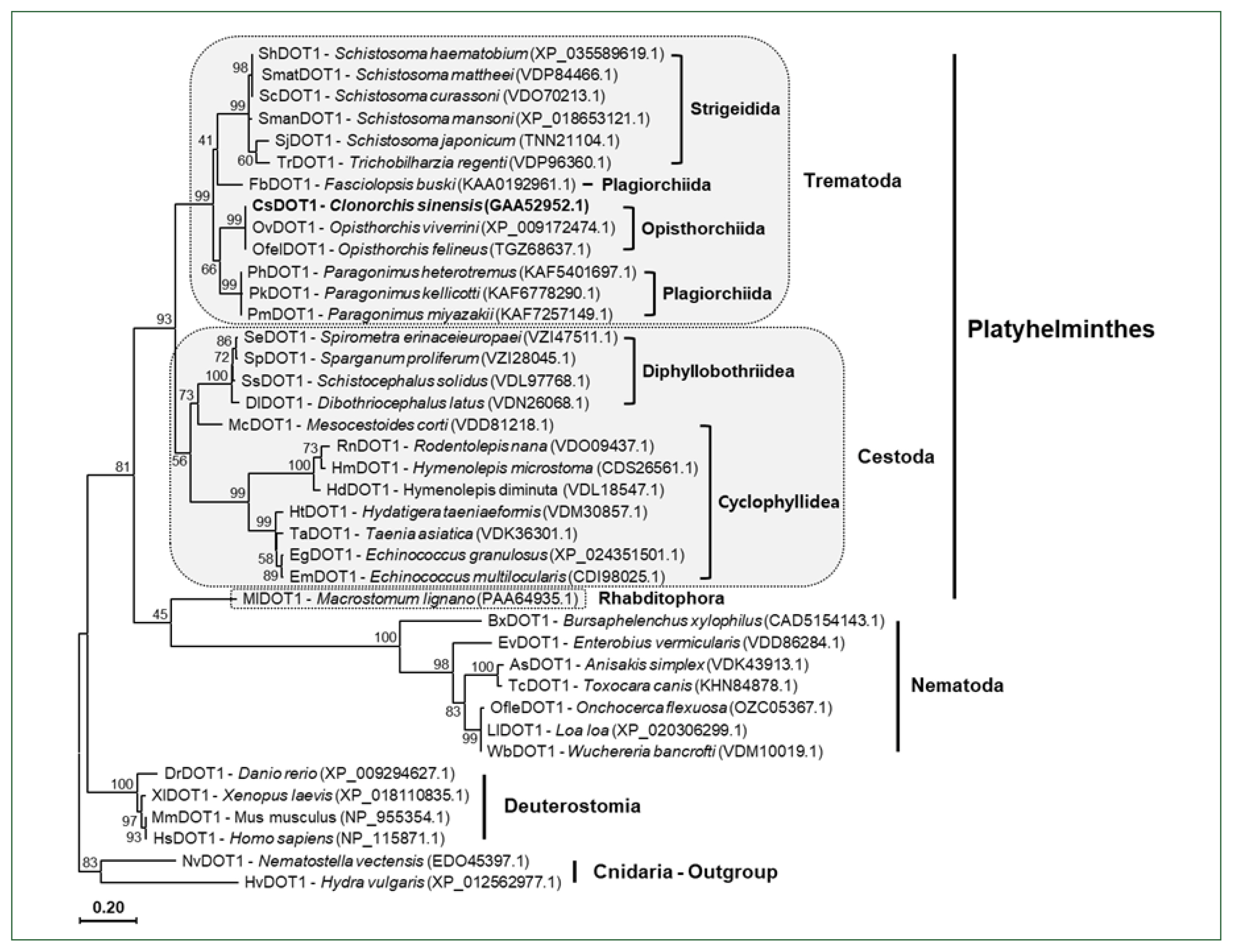
A phylogenetic tree of platyhelminth and nematode DOT1. The amino acid sequences of the DOT1 domain were isolated from some representative platyhelminth and nematode species including C. sinensis. The sequences were used in the phylogenetic analysis, together with those of deuterostomian and cnidarian orthologs, using the PhyML program. The tree was rooted with the cnidarian DOT1s. Numerals at branching nodes indicate the bootstrap values (1,000 replicas).
Methylated histones in C. sinensis adults
The aa sequences of Clonorchis histones H3 and H4 were isolated from GenBank database by BLAST searches using the sequences of human histones H3.1 (NP_003522.1) and H4 (NP_003486.1). The aa sequences were highly conserved among these fluke and human homologs (identity values >98.0%), except for that of the C. sinensis histone H3.1 (KAG5455013.1, 69.2%) (Supplementary Fig. S1). Consistent with the tight aa conservation, pan-antibodies against histone H3 and its methylated derivatives were highly reactive with the nuclear extracts of C. sinensis adults (Supplementary Table S1; Supplementary Fig. S2). Using these antibodies, the histological distribution of the methylated histones was examined across the whole body of the fluke. The anti-histone H3 rabbit antibody stained the nuclei in the parenchymal region and sexual organs of C. sinensis adults, as well as those in intrauterine eggs. Antibodies against H3K4me3, H3K27me3, and H3K79me3 showed staining patterns similar to that of the anti-H3 antibody, whereas the relative intensity of the positive signal differed slightly among the antibodies and tissues/organs. However, the specific targets of anti-H3K36me3 and H4K20me3 antibodies were largely detected in the vitellaria and intrauterine eggs. Proteins in the nuclei of ovaries, testes, and seminal receptacle reacted minimally with these antibodies. Finally, a weak positive signal was observed in the intrauterine eggs but not in the other tissues/organs, in the immunohistochemical staining images obtained with anti-H3K9me3 antibody (Fig. 5).
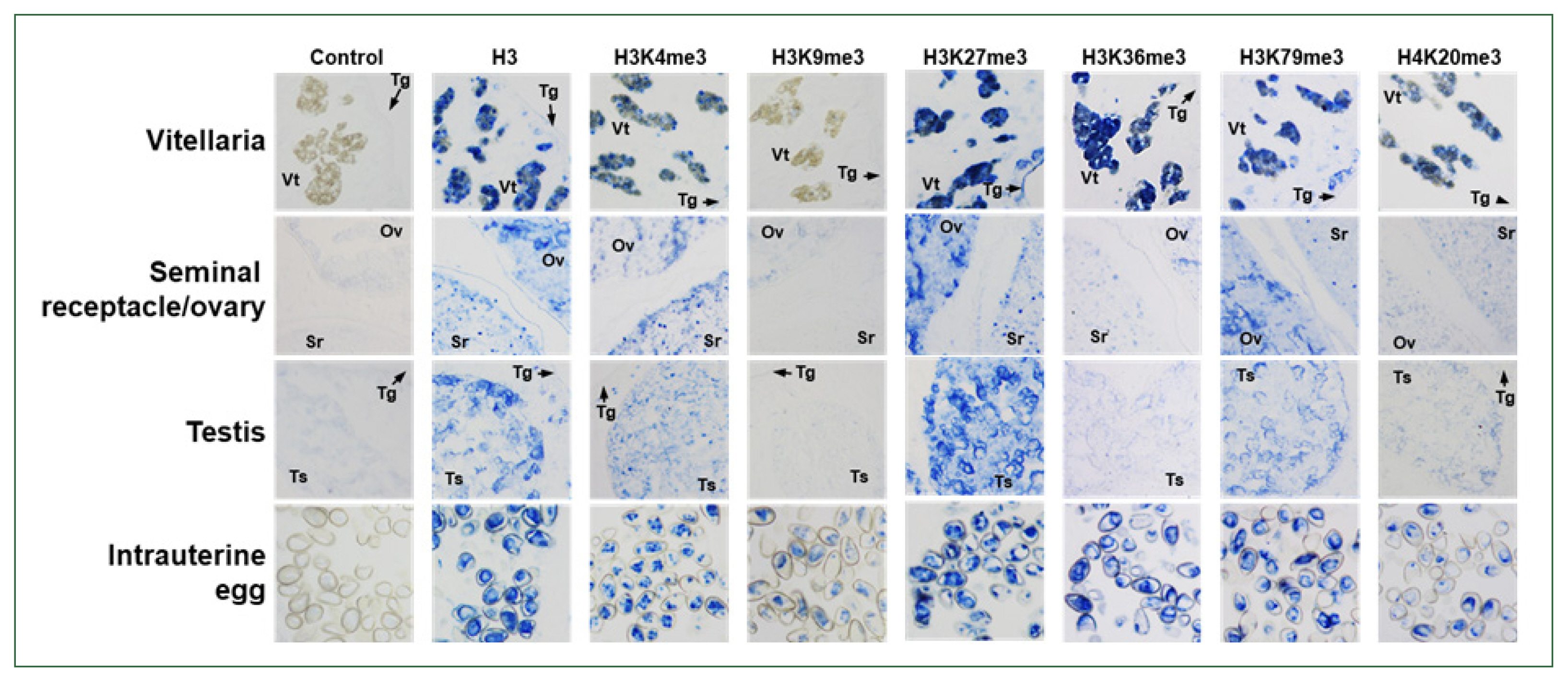
Immunological detection of methylated histones in Clonorchis sinensis adults. The whole-body sections of C. sinensis adults (42-day-old) were reacted with antibodies specific to histone H3 and its methylated derivatives, as indicated on the top, and the positive reactions were enzymatically stained with the HIGHDEF blue chromogen. Rabbit normal serum was used in the control staining reaction. Ov, ovary; Sr, seminal receptacle; Tg, tegument; Ts, testis; Vt, vitelline follicle.
Temporal profiling of SET-HKMT gene expressions
Six Clonorchis SET-HKMT genes (CsSET-HKMT: CsSETD4 for GAA51355, CsPRDM9 for GAA52155, CsEZH for GG55462, CsNSD1/2 for GAA51760, CsSUV420H for GAA53884, and CsSETD8 for GAA4871) were arbitrarily selected based on their phylogenetic positions (solid dots in Fig. 2), and their temporal expression profiles were examined in C. sinensis via the RT-qPCR analysis. The relative expression levels of these genes gradually increased during the development/maturation of the worm from metacercaria to 15-day-old worm, except for CsPRDM9 and CsSUV420H, expressions of which were reduced in 6-day-old juveniles compared to those in metaceracariae, and then significantly decreased in 25-day-old worms (P<0.001; Fig. 6). The relative expression of CsNSD1/2 and CsSETD8 was further reduced in 84-day-old worms, similar to other functional genes such as C. sinensis tyrosinases (CsTyrs) and myoglobins (CsMbs) [25,29]. However, the transcriptional activity of the other 3 genes was significantly restored in the later stages of C. sinensis adults (P values for CsSETD4, CsPRDM9, and CsSUV420H<0.01).
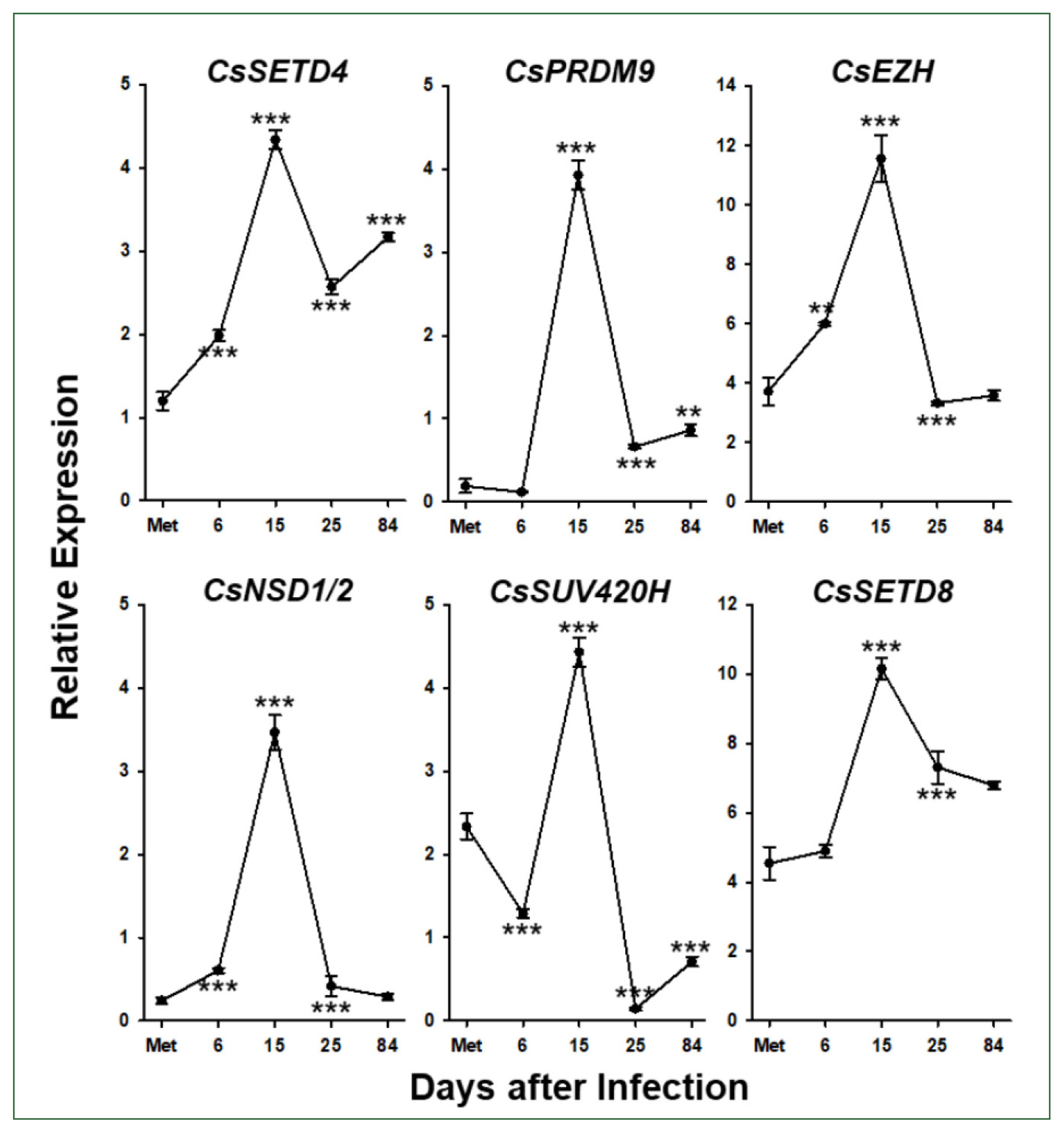
Temporal expression profiles of Clonorchis sinensis histone lysine methyltransferase (CsHKMT) genes. Expression profiles of the C. sinensis genes were determined using quantitative reverse transcription PCR, in association with the development/maturation of the liver fluke from metacercaria to 84-day-old worm. The calculations are based on independent technical triplicates (n=3, mean±standard deviation). The significance of the expression changes at each stage was statistically estimated by comparing the gene expression levels with those of its direct previous stage using the Student’s t test. **P<0.01, ***P<0.001.
Effects of HKMT inhibitors on functional gene expression
In our previous studies, the temporal expressions of C. sinensis DNA methyltransferase (CsDNMT2) and methyl-CpG binding protein (CsMBD1 and 2) genes showed patterns similar to that of CsEZH (Supplementary Fig. S3). Taken together with the present results, these expression patterns suggest that epigenetic gene regulation via DNA and/or histone methylation increases in C. sinensis with age. To test this hypothesis, the changes in functional gene expressions (CsSET-HKMTs, CsMBDs, CsDNMT2, CsTyrs, and CsVits) were examined in 42- and 140-day-old C. sinensis worms treated with the methyl transferase inhibitors SGI-1027 and DZNep for 5 days. As shown in Fig. 7, the transcriptional activities of these C. sinensis genes were significantly affected in the 140-day-old worms (P values<0.05). Overall, SGI-1027 induced these functional genes, but DZNep down-regulated them in the older worms, whereas the degrees of the responsive changes differed substantially depending on the chemicals and genes (Fig. 7). In 42-day-old worms, both chemicals strongly activated the CsMBD2 and CsDNMT2 genes. However, CsTyr and CsVit expressions were not significantly affected by SGI-1027 or DZNep in the younger worms (Fig. 8). Interestingly, the individual genes encoding SET-HKMTs responded differently to these inhibitors in both younger and older worms (Figs. 7, 8).
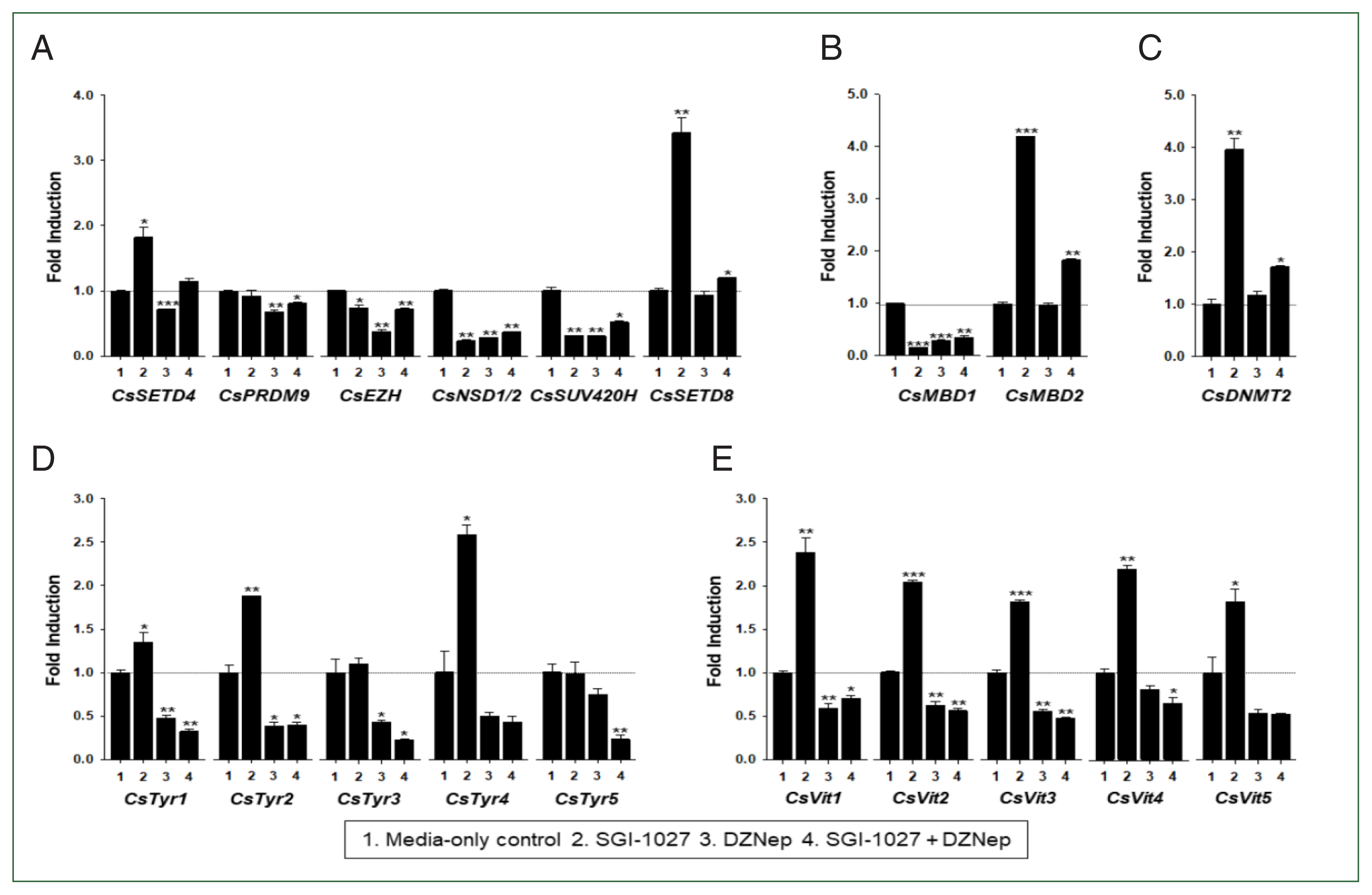
Effect of methyltransferase inhibitors on functional gene expressions in 140-day-old Clonorchis sinensis adults. Live C. sinensis worms were incubated in the RPMI-1640 medium supplemented with SGI-1027 (5 μm) and/or 3-deazaneplanocin A (DZNep, 0.5 μm) in a 5.0%-CO2 incubator. After a 24 h incubation, total RNAs were extracted from the worms and used in quantitative reverse transcription PCR with primer sets specific to each of the C. sinensis genes. (A) HKMT genes; (B) methyl-CPG; (C) DNMT genes; (D) Tyrosinase genes; (E) eggshell precursor protin genes. Fold increases in the globin gene expression in each of the experimental groups were calculated against the media-only control group (n=3, mean±standard deviation). *P<0.05, **P<0.01, ***P<0.001.
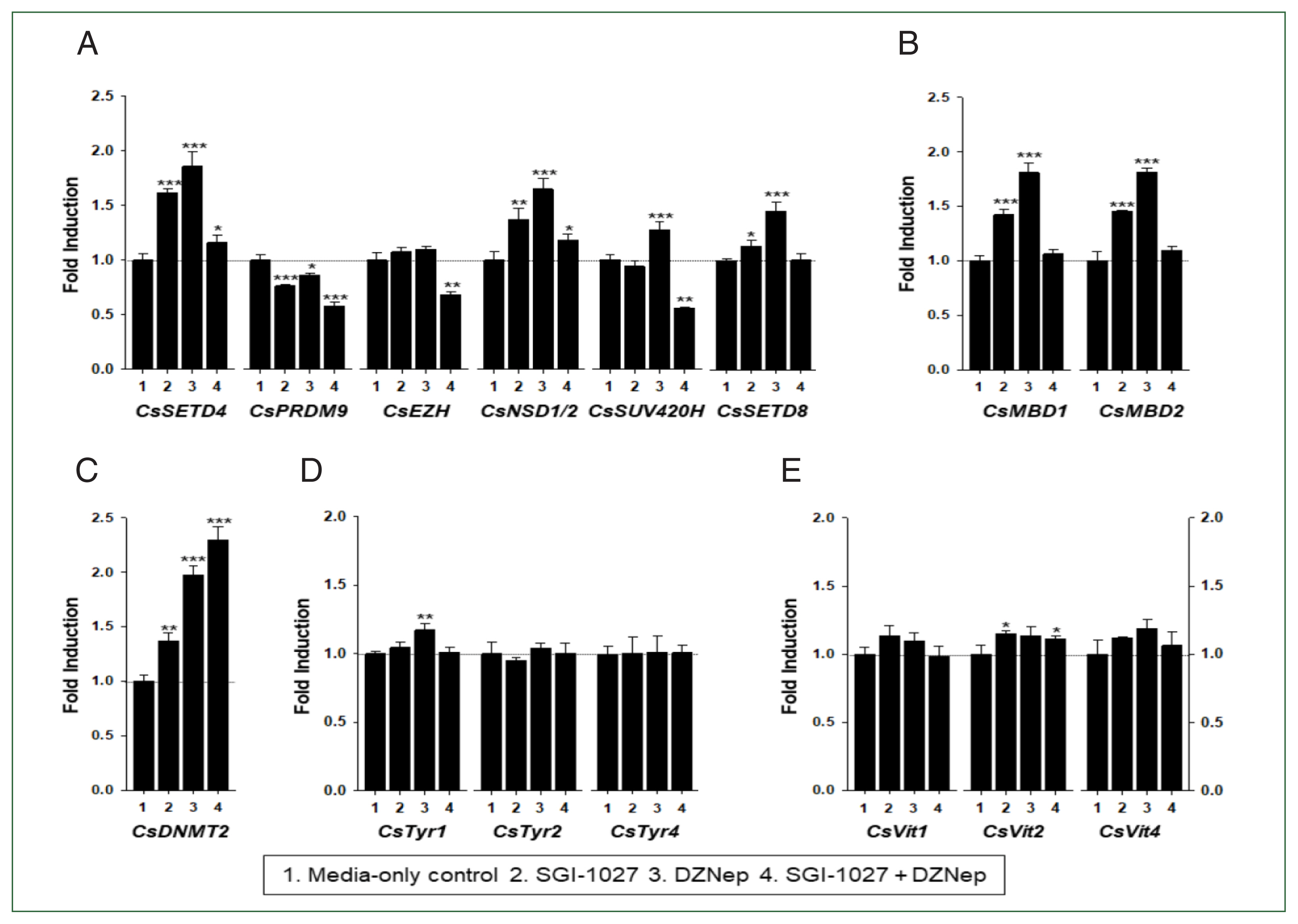
Effect of methyltransferase inhibitors on functional gene expressions in 42-day-old Clonorchis sinensis adults. Live C. sinensis worms were incubated in the RPMI-1640 medium supplemented with SGI-1027 (5 μm) and/or 3-deazaneplanocin A (DZNep, 0.5 μm) in a 5.0% CO2 incubator. After a 24 h incubation, total RNAs were extracted from the worms and quantitative reverse transcription PCRs were performed using primer sets specific to each of the C. sinensis genes. (A) HKMT genes; (B) methyl-CPG; (C) DNMT genes; (D) Tyrosinase genes; (E) eggshell precursor protin genes. Fold increases in the globin gene expression in each of the experimental groups were calculated against the media-only control group (n=3, mean±standard deviation). *P<0.05, **P<0.01. ***P<0.001.
Discussion
Since DNMT2 was shown to be an RNA methyltransferase [30] and platyhelminths belong to the so-called DNMT2-only organisms [10], the presence of DNA methylation-related epigenetic machinery and/or its practical implications in gene regulation have been highly disputable for a long time in the lower animals. However, recent studies have provided multiple evidences revealing that the epigenetic machinery effectively operates in the lower animal group [14,21,31]. In addition to the previously described 5-methylcytosines [21], this study showed that methylated histone variants are intimately involved in the epigenetic regulation of C. sinensis genes. For the differential histone methylation, genes encoding most of the HKMT family members, which have been widely identified across animal taxa, are preserved in the liver fluke and other platyhelminth genomes, while some genes were specifically expanded in a series of donor organisms. The temporal expression profiles of CsHKMT and other functional genes suggest that histone methylation is deeply involved in epigenetic pathways that contribute to age-associated phenotypes, such as reduced fecundity in C. sinensis. This aging process may be reversed by inhibiting CsHKMT activities.
The mammalian SET-HKMT family can be categorized into approximately 6 subfamilies (SET1 for H3K4, SUV39 for H3K9, SET2 for H3K36, EZH for H3K27, SMYD for H3K4, and PRDM for H3K9) and an extra group comprising multiple heterogeneous proteins based on structural and enzymatic characteristics [22]. Most of these subfamily members have specific orthologous and/or biochemical counterparts in plants [32], demonstrating that the protein family may have varied during an early stage of metazoan evolution before divergence of animals and plants. Parasitic helminths examined in this study have well preserved all these subfamilies: SETD1, MLL, and MLL3 lineages in SET1; SETMAR, SETB1, and SUV39H1/2 lineages in SUV39; NSD1-3 and SETD2 lineages in SET 2; EZH lineage in EZH; SMYD4/5 lineages in SMYD; PRDM lineage in PRDM; and the other heterogeneous SET4/8, SUV420H, and MLL5 lineages (Fig. 2). A single gene encoding the non-SET HKMT protein DOT1, which is responsible for H3K79 methylation, was also well conserved in each parasite genome (Figs. 3, 4). The high gene dosages were in contrast to that of DNMT gene. Platyhelminth genomes preserved only a single DNMT2 homolog but no DNMT1 or DNMT3 homologs [21].
The life cycle of C. sinensis comprises 7 distinct developmental stages involving 3 sequential hosts: egg; miracidium, sporocyst, redia, and cercaria in freshwater snail; metacercaria in freshwater fish; and adult in mammal [33]. The worms at discrete stages exhibit great differences in morphology, size, structure, and tissue/organ composition. Therefore, it can be postulated that the parasite is highly dependent on methylated histone-mediated epigenetic processes for stage-specific gene expression profiles on an identical genetic background, as has recently been examined in S. mansoni [14]. Consistent with the large enzymatic pool, diversely methylated histones were identified in whole-worm sections of C. sinensis adults (Fig. 5). In general, the methylated forms of H3K4 and H3K79 are associated with the transcriptional activation of relevant genes, whereas those of H3K9, H3K27, and H3K36 are closely linked to euchromatic and/or heterochromatic silencing. In contrast to the mono- and di-methylated forms, the tri- methylated form of H4K20 also serves as a silencing marker [7]. The relative densities of these epigenetic markers were substantially different among the C. sinensis tissues/organs. H3K79me3 and H3K27me3 are likely the major transcriptional activator and repressor, respectively, in the vitellaria, ovaries, and testes. The repressive H3K36me3 was also highly enriched in the vitellaria. The activating H3K4me3 and repressive H4K20me3 were moderately detected in these organs. However, all histone markers examined in this study were expressed at relatively high levels in intrauterine eggs (Fig. 5).
The genome-wide methylation status of DNAs and histones is significantly altered by reversible demethylation-methylation events throughout the development and organogenesis in animals [34]. The major epigenetic markers, especially H3K4me3 and H3K27me3, also fluctuate highly in S. mansoni in accordance with developmental progress [14]. Although the methylome changes were not investigated following the development in C. sinensis in this study, it was apparent that the genome-wide densities of methylated histones in intrauterine eggs differ from those in other tissues/organs in C. sinensis adults. The fertilized ovum (i.e., zygote) of C. sinensis and S. mansoni is enclosed in a proteinaceous shell within the ootype and embryonate into the miracidium while passing through the uterine tube. Finally, fully embryonated egg is released from the uterine pore and hatches in fresh water (S. mansoni) or in the first snail host (C. sinensis) [35]. The oscillating genome-wide frequency of H3K27me3 during schistosome development was the lowest at the miracidium stage, probably due to global demethylation after the fusion of gamete cells [14]. However, the densities of the diversely methylated histones, including H3K27me3, were considerable in the embryonated eggs of C. sinensis. Considering that the genome-wide frequencies of methylated histones increase according to the maturation and/or aging of donor organisms [34], this observation may reflect differentially operating epigenetics between the 2 trematode parasites. Alternatively, this fact possibly results from different degrees of maturation, i.e., developing vs. fully developed miracidia. Further investigation on the different maturation stages of miracidium may help address this issue.
The fertility rate of the hermaphroditic C. sinensis gradually declines to a very low level as the worm ages in the definitive hosts after reaching a peak [36]. The temporal expressions of Clonorchis genes, such as CsTyrs and CsMbs, the protein products of which play key roles in sexual reproduction, appears to be synchronized with the change in fecundity [25,29]. However, the expression of genes encoding epigenetic writers (HKMTs and DNMT) and readers (MBDs) significantly increased in later adulthood (Fig. 6 and Supplementary Fig. S3). Therefore, it could be postulated that the fecundity reduction is attributable to the down-regulation of essential genes corresponding to the increasing frequencies of epigenetic markers. This presumption was, at least in part, supported by observations showing that functional gene expressions are differentially regulated by histone/DNA methylation depending on the age of C. sinensis adults (Figs. 7, 8). In 42-day-old worms, epigenetic tool genes were likely repressed by DNA and histone methylation, but those related to eggshell formation were not under the epigenetic control (Fig. 8). In contrast, eggshell-related genes and some epigenetic tool genes were epigenetically silenced mostly by DNA methylation in 140-day-old worms. The significant down-regulation of HKMT and MBD1 by treatment with methyltransferase inhibitors in the older worms further revealed that epigenetic activators, such as H3K4me3, are intimately involved in the up-regulation of epigenetic tool genes (Fig. 7). S-adenosylhomocysteine (SAH) hydrolase-inhibiting DZNep and SAM-competitive SGI-1027 are known to be specific to EZH2 [37] and DNMTs [38], respectively. However, these molecules may act as pan-methyltransferase inhibitors, as most cellular methyltransferases use SAM as a methyl donor and are inhibited by SAH, which is generated by the methylation reaction and hydrolyzed into adenosine and homocysteine by SAH hydrolase [39]. Therefore, the modulated gene expressions by the chemical treatments are likely the sum of broad methyltransferase inhibition rather than restriction to a specific CsEZH or CsDNMT2 blockade. Nevertheless, our data clearly demonstrate the significant role of the epigenetic machinery in the temporal regulation of C. sinensis genes, especially those involved in sexual reproduction.
Methylated DNA and histones are highly interrelated and depend mechanistically on each other for the epigenetic regulation of gene expression and/or maintenance of chromosomal integrity [40]. Most multicellular eukaryotes express 3 DNMTs that generate (DNMT3a and DNMT3b) and maintain (DNMT1) the genome-wide 5mC methylomes. However, the genomes of C. sinensis and other platyhelminths encode only a single DNMT protein homologous to the mammalian DNMT2 [21] and the DNMT2-like protein seems to provide enzymatic activity responsible for the generation and/or maintenance of DNA methylomes [21,31]. This situation is highly contrasted to the well-preserved HKMT dosages in these invertebrates (Figs. 2, 4). The reduced dosage of DNMT gene would directly contribute to the low frequency of genome-wide 5mC in C. sinensis and S. mansoni [21,31]. However, whether the imbalanced genetic dosages of DNMT and HKMT in the DNMT2-only organisms influence on taking initiative during epigenetic process remains unclear. Locus-specific methylome maps previously obtained in S. mansoni were constructed using chromatins extracted from whole worm bodies [14,31]. Cells comprising various tissues/organs in multicellular organisms are heterogeneous in terms of epigenetic modifications [34]. Consequently, cell- and tissue-specific methylome patterns and their implications in the regulation of nearby genes in parasitic trematodes have not yet been defined. Future investigations with homogeneous cell populations should address issues regarding the detailed interrelationship between DNA and histone methylation, and the tissue-specific methylome construction in association with the development and/or maturation stages in the DNMT2-only organisms.
Supplementary Information
Comparison of primary structures of histones H3 (panel A) and H4 (panel B) between Clonorchis sinensis and human. The amino acid (aa) sequences of histone H3 and H4 paralogs were retrieved from the GenBank databases and were aligned against one another for comparison of their primary structures. The degrees of the aa conservations are highlighted using different shades of grey, and gaps represented by dots were introduced in the alignment to increase the similarity values. Solid lines under the alignments represent the histone fold domains conserved in eukaryotic histones H3 and H4 (Postberg et al., BMC Evolutionary Biology 2010, 10:259). The Lys residues majorly targeted by histone lysine methyltransferases for methylation are indicated by solid dots. The percentage values in the parentheses following proteins names are similarity values calculated against Hs_H3.1 or Hs_H4.
Detection of methylated histone derivatives in the cellular protein extracts of Clonorchis sinensis. The cytosolic (C) and nuclear (N) extracts of C. sinensis adults were reacted with antibodies specific to the histone derivatives after being resolved via 15.0% SDS-PAGE. The protein extracts were similarly prepared from human HeLa cells and rat liver parenchyma and used in the experiment.
Temporal expressions of Clonorchis sinensis CsEZH (protein accession number GAA55462), CsMBD1 (GAA50068), CsMBD2 (GAA57483), and CsDNMT2 (GAA54195) genes. The gene expression profiles were examined by the quantitative reverse transcription PCR analysis of total RNAs, which were extracted from C. sinensis worms at different maturation stages from 4- to 140-day-old. The calculations are based on independent technical triplicates (n = 3, mean ± standard deviation). The statistical significance of the expression changes (P values of the Student’s t test) at the latest stage, compared to those of its direct previous stage, is indicated in the graph.
Supplementary Table S1. Antibodies used for the detection of Clonorchis sinensis histone and its methylated derivatives.
Supplementary Table S2. Primers used in this study.
Supplementary Table S3. SET-HKMT family proteins idnetified in the proteomic database of Schistosoma mansoni.
Supplementary Table S4. SET-HKMT family proteins idnetified in the proteomic database of Echinococcus granulosus.
Supplementary Table S5. SET-HKMT family proteins idnetified in the proteomic database of Hymenolepis microstoma.
Supplementary Table S6. SET-HKMT family proteins idnetified in the proteomic database of Schmidtea mediterranea.
Supplementary Table S7. SET-HKMT family proteins idnetified in the proteomic database of Globodera pallida.
Acknowledgment
This work was supported by the Basic Science Research Programs of the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2017R1D1A1B03 028355) and by the Gachon University research fund of 2018 (GCU-2018-0329) to YAB.
Notes
The authors declare no conflict of interest related to this study.
Conceptualization: Sohn WM, Bae YA
Data curation: Bae YA
Formal analysis: Park MJ
Funding acquisition: Bae YA
Investigation: Park MJ
Methodology: Park MJ
Project administration: Bae YA
Resources: Sohn WM
Supervision: Bae YA
Validation: Sohn WM, Bae YA
Visualization: Bae YA
Writing – original draft: Bae YA
Writing – review & editing: Sohn WM, Bae YA