Abstract
Toxoplasma gondii is an intracellular parasite that causes severe disease when the infection occurs during pregnancy. Adenosine is a purine nucleoside involved in numerous physiological processes; however, the role of adenosine receptors in T. gondii-induced trophoblast cell function has not been investigated until now. The goal of the present study was to evaluate the intracellular signaling pathways regulated by adenosine receptors using a HTR-8/SVneo trophoblast cell model of T. gondii infection. HTR8/SVneo human extravillous trophoblast cells were infected with or without T. gondii and then evaluated for cell morphology, intracellular proliferation of the parasite, adenosine receptor expression, TNF-α production and mitogen-activated protein (MAP) kinase signaling pathways triggered by adenosine A3 receptor (A3AR). HTR8/SVneo cells infected with T. gondii exhibited an altered cytoskeletal changes, an increased infection rate and reduced viability in an infection time-dependent manner. T. gondii significantly promoted increased TNF-α production, A3AR protein levels and p38, ERK1/2 and JNK phosphorylation compared to those observed in uninfected control cells. Moreover, the inhibition of A3AR by A3AR siRNA transfection apparently suppressed the T. gondii infection-mediated upregulation of TNF-α, A3AR production and MAPK activation. In addition, T. gondii-promoted TNF-α secretion was dramatically attenuated by pretreatment with PD098059 or SP600125. These results indicate that A3AR-mediated activation of ERK1/2 and JNK positively regulates TNF-α secretion in T. gondii-infected HTR8/SVneo cells.
-
Key words: Toxoplasma gondii, adenosine A3 receptor, TNF-α, HTR8/SVneo trophoblast cell, MAPK
INTRODUCTION
Toxoplasma gondii is an obligate intracellular protozoan parasite that can infect many vertebrate animals, including humans, and is highly prevalent worldwide [
1]. Although
T. gondii infection in humans is frequently asymptomatic, it can lead to severe disease in immunocompromised patients and congenitally infected children, leading to several manifestations, such as retinochoroiditis and miscarriage during the first trimester of pregnancy [
2]. Host protection against
T. gondii infection results from a complex cell-mediated immune response involving inflammatory cells, lymphocytes and macrophages, which is characterized as a T helper type 1 (Th1)-immune response with prominent production of interferon (IFN)-γ, tumor necrosis factor (TNF)-α and interleukin (IL)-1β [
3].
Disorders due to congenital
T. gondii infection likely involve both cellular and molecular changes in the placenta.
T. gondii is known to infect all nucleated host-cells and can trigger host-cell apoptosis [
4]. In pregnant mice infected by
T. gondii, increased trophoblast apoptosis was observed and was associated with increased FAS expression in trophoblast cells and IFN-γ and TNF-α in decidual cells [
5].
T. gondii infection-induced apoptosis was also observed in femur bone marrow cells of mice and was associated with increased TNF-α expression [
6]. TNF-α, a multifunctional cytokine, has been detected in many tissues including ovary, oviduct, uterus, and placenta and is expressed in embryonic tissues. For many years, TNF-α was primarily considered to be a cytokine involved in triggering immunological pregnancy loss and as a mediator of various embryopathic stresses [
7].
Adenosine is a potent immunomodulatory biomolecule that is produced by the ectoenzymes nucleoside triphosphate dephosphorylase (CD39) and ecto-5′-nucleotidase (CD73), which are highly expressed by several cell types, including leukocytes, during stress, injury, and infection [
8]. Extracellular adenosine levels increase in response to hypoxia, ischemia and inflammation, preventing tissue damage during instances of cellular stress or injury [
9]. The effects of adenosine are mediated via 4 adenosine receptor (AR) subtypes: A
1AR, A
2AAR, A
2BAR, A
3AR [
10]. Of these, A
2AAR is recognized as mediating major adenosine anti-inflammatory activity. Iriyama et al.[
11] revealed that a local increase of adenosine in the placenta is sufficient to trigger key features of preeclampsia using mouse models, and adenosine was identified as one of pathogenic factors for preeclampsia. A2B receptor activation has been shown to blunt trophoblast migration, possibly as a result of reduced activation of the ERK1/2 and SAPK/JNK signaling pathway and lower proMMP-2 and VEGF levels, which are crucial for trophoblast function [
9]. These observations suggest the possible involvement of adenosine receptors in placental developmental processes.
Although adenosine receptor activity is important in the immune response against
T. gondii during gestation, the role of adenosine receptors in
T. gondii-infected human extravillous trophoblast cells remains unclear. HTR8/SVneo cells are widely used as a model of human extravillous trophoblast cells to evaluate the migration and proliferation processes of extravillous trophoblast cells [
12] and better understand the factors involved during preeclampsia [
13]. For this reason, HTR8/SVneo cells were used as to study
T. gondii infection in the present study.
The goal of the present study was to evaluate the functional role of adenosine receptors using a HTR-8/SVneo trophoblast cell model of T. gondii infection, we evaluated T. gondii-mediated alterations in HTR8/SVneo cell morphology, intracellular proliferation of the parasite, adenosine receptor expression, TNF-α production and mitogen-activated protein kinase (MAPK) signaling pathways triggered by A3AR in HTR8/SVneo cells.
MATERIALS AND METHODS
Cell culture and parasite infection
The human extravillous trophoblast cell line HTR8/SVneo was purchased from the American Tissue Culture Collection (Manassas, Virginia, USA). Briefly, these cells were cultured in RPMI 1640 medium (Hyclone, Waltham, Massachusetts, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotic-antimycotic (all from Gibco, Grand Island, New York, USA) in a humidified incubator at 37°C under an atmosphere with 5% CO2. HTR8/SVneo cells were infected with T. gondii at a multiplicity of infection (MOI) of 10 for 4, 8, and 24 hr. Then the cell morphology, viability, adenosine receptor family expression, TNF-α production and activation of MAPK signaling pathways were evaluated. To evaluate the role of A3AR in T. gondii-induced MAPK activation and TNF-α production, HTR8/SVneo cells that had been incubated for 48 hr after transfection with control siRNA or A3AR-specific siRNAs were infected with T. gondii at an MOI of 10 for 24 hr and then MAPK activation and TNF-α secretion levels were assessed.
In vitro cultivation of Toxoplasma gondii
Tachyzoites of RH and GFP-RH (constitutively express green fluorescent protein) strains of T. gondii were maintained in ARPE-19 cells under an atmosphere with 5% CO2 and 37°C. Infected cells were scraped, forcibly passed through a 27-gauge needle, and centrifuged at 1,350×g for 10 min using Percoll (Sigma, St. Louis, Missouri, USA) to pellet the parasites.
The human RPE cell line ARPE-19 was purchased from the American Tissue Culture Collection (Manassas, Virginia, USA). The cells were routinely grown in Dulbecco’s modified Eagle’s medium/F12 (Hyclone) supplemented with 10% heat-inactivated fetal bovine serum and antibiotic–antimycotics (all from Gibco). The cells were cultured at 37°C under an atmosphere with 5% CO2 and passaged every 3–4 days. ARPE-19 cells were used between passages 4 and 8 in the present study.
Immunofluorescence microscopy
HTR8/SVneo cells were seeded onto coverslips in 12-well plates at a density of 2×104 cells/well and incubated for 24 hr. The cells were then mock-infected or infected with the GFP-RH strain of T. gondii at a multiplicity of infection (MOI) of 10 for 4, 8, and 24 hr. Subsequently, the cells were washed with Hank’s balanced salt solution (HBSS) and fixed with freshly prepared 4% paraformaldehyde for 1 hr at room temperature. After being washed 5 times with PBS containing 0.3% Triton X-100 (PBS-T) for 10 min, the cells were incubated with a primary antibody against α-tubulin for 2 hr at room temperature. The cells were washed to remove excess primary antibody and then incubated with an anti-mouse Alexa Fluor 647 secondary antibody for 2 hr. After mounting the samples with VECTASHIELD HardSet antifade mounting medium with DAPI (Vector Laboratories, Burlingame, California, USA), fluorescence images were acquired using a laser scanning confocal microscope (Leica TCS SP5 II).
Cell viability assay
HTR8/SVneo cells were either mock-infected or infected with T. gondii at an MOI of 10 for 4, 8 or 24 hr, after which cell viability was estimated using a CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (Promega, Madison, Wisconsin, USA) following the manufacturer’s instructions.
Western blot analysis
After washing HTR8/SVneo cells with phosphate-buffered saline (PBS), proteins were isolated using ice-cold RIPA buffer (Thermo Scientific, Waltham, Massachusetts, USA) with complete protease inhibitor cocktail (Roche, Basel, Switzerland) for 30 min on ice. After centrifugation at 14,000×g for 15 min at 4°C, the supernatants were collected, and equal amounts of protein from each sample were separated by SDS-PAGE and then transferred to polyvinylidene difluoride membranes. The membranes were blocked in Tris-buffered saline (20 mM Tris and 137 mM NaCl, pH 7.6) containing 0.1% Tween-20 (TBST) and 5% skim milk. After being washed once with TBST, the membranes were incubated overnight at 4°C with the primary antibodies diluted in TBST supplemented with 5% bovine serum albumin. Primary antibodies against the following proteins were used: adenosine A1-R (A1AR), adenosine A2A-R (A2AAR), adenosine A2B-R (A2BAR), adenosine A3-R (A3AR), and α-tubulin (all from Santa Cruz Biotechnology); and phospho-p38 MAPK, p38 MAPK, phospho-ERK1/2, ERK1/2, phospho-JNK, and JNK (all from Cell Signaling Technology Inc., Danvers, Massachusetts, USA). Following 3 consecutive washes in TBST, the membranes were incubated for 90 min with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Jackson ImmunoResearch Laboratories), diluted 1:5,000 with incubation buffer, as described above. Subsequently, the membranes were soaked with Immobilon Western Chemiluminescent HRP Substrate (Jackson ImmunoResearch Laboratories), and chemiluminescence was detected with a Fusion Solo System (Vilber Lourmat, Collegien, France). Band intensity was quantified using ImageJ (NIH, Bethesda, Maryland, USA), and the results were normalized to α-tubulin protein levels and expressed as fold-changes compared to the control group.
RNA extraction and quantitative reverse transcription PCR (RT-qPCR)
Total cellular RNA was extracted using TRIzol Reagent (Invitrogen Life Technologies, Carlsbad, California, USA). RNA (2 μg) was reverse transcribed in a final volume of 20 μl using Superscript II reverse transcriptase (Invitrogen Life Technologies), as specified by the manufacturer, and then used in RT-qPCR analyses with specific primer sets (
Table 1). RT-qPCR was conducted using an ABI 7500 FAST System (Applied Biosystems, Carlsbad, California, USA) in 20-μl reactions containing cDNA (100 ng) with SYBR-Premix Ex Taq II (Takara Bio Inc., Otsu, Japan). The hypoxanthine phosphoribosyltransferase 1 (HPRT1) gene was amplified for normalization of the cDNA amount used in RT-qPCR. Reactions were performed in triplicate, and the data were analyzed using the 2
−ΔΔCt method.
HTR8/SVneo cells were mock-infected or infected with the RH strain of T. gondii at an MOI of 10 for 4, 8, and 24 hr. The supernatants from the mock-or T. gondii-infected HTR8/SVneo cells were collected in triplicate, and TNF-α levels were measured using commercially available TNF-α ELISA kits following the manufacturer’s instructions (R&D System, Minneapolis, Minnesota, USA). The cytokine concentrations in the samples were calculated from standard curves obtained using recombinant cytokines.
siRNA transfection
Cells were transfected with siRNA duplexes specific for human A3AR (Santa Cruz Biotechnology) using Lipofectamine RNAiMAX (Life Technologies, Carlsbad, California, USA) following the manufacturer’s protocol. Briefly, the cells were seeded into 6-well plates, grown for 24 hr (70% confluence), and then transfected with 20 nM adenosine A3-R siRNA or negative control siRNA (Santa Cruz Biotechnology Inc., Santa Cruz, California, USA) for 48 hr. Subsequently, the cells were pre-infected with the RH strain of T. gondii at an MOI of 10 for 24 hr, and knockdown efficiency was determined by western blot analysis.
Inhibition of MAPK activation
To confirm the roles of p38, ERK1/2, and JNK in the T. gondii-induced production of TNF-α by HTR8/SVneo cells, we performed experiments using inhibitors of p38 MAPK (SB203580), ERK1/2 (PD098059), and JNK (SP600125). HTR8/SVneo cells were pretreated with 30 μM SB203580, PD098059 or SP600125 for 1 hr followed by infection with T. gondii at an MOI of 10 for a further 24 hr. Then, the cell culture supernatants were collected, and TNF-α secretion was assessed as described above.
Statistical analyses
All assays were performed at least 3 times in triplicate. The data are presented as means±standard deviation (SD). Statistical analysis of the data was performed using unpaired, 2-tailed Student’s t-tests with Bonferroni adjustment or ANOVA for multiple comparisons. A P-value less than 0.05 was considered to indicate statistical significance.
RESULTS
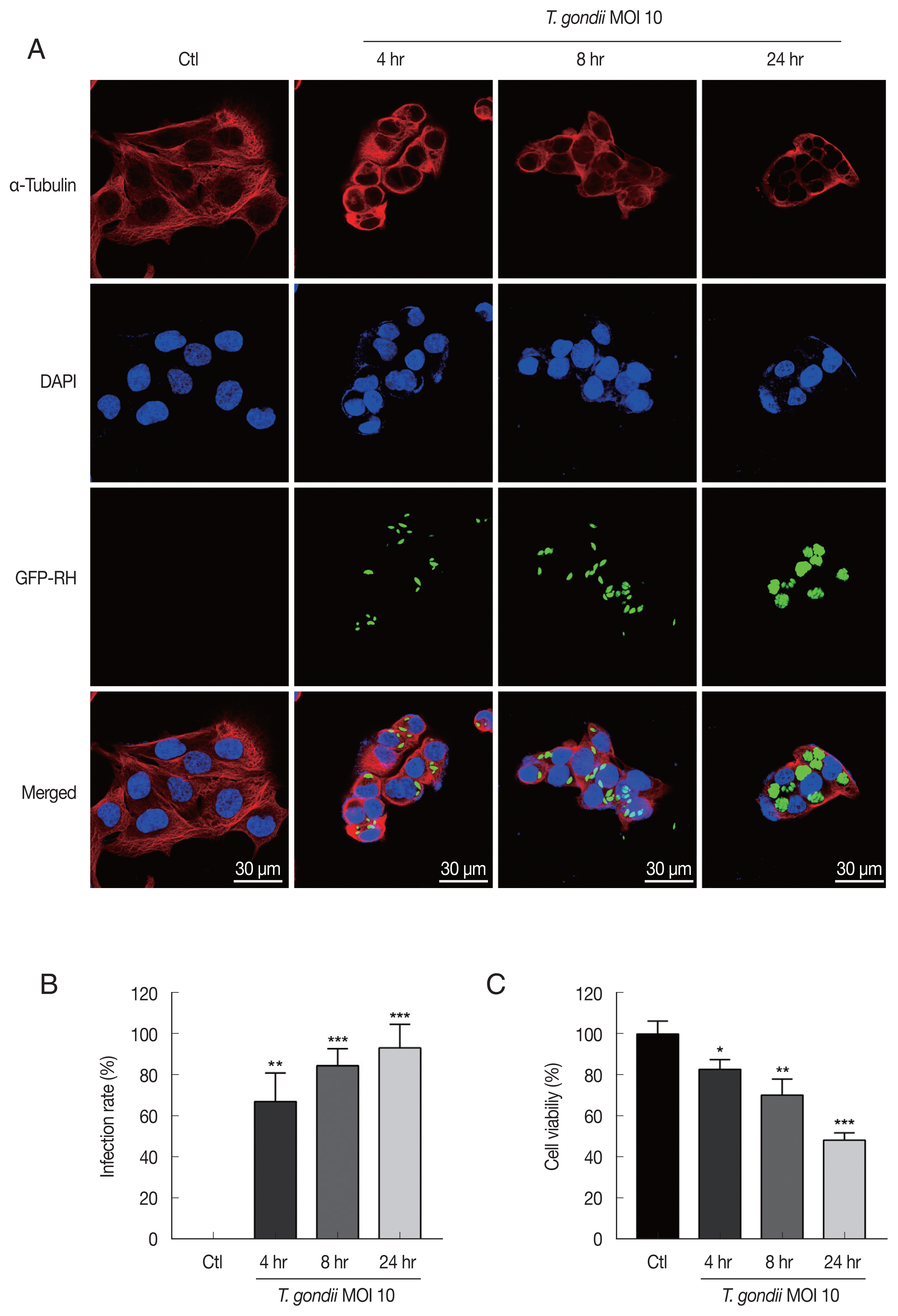
T. gondii infection induced cytoskeletal changes in HTR8/SVneo cells and reduces cell viability
For detecting morphologic changes, the integrity of the microtubule network was assessed by immunofluorescence microscopy using an α-Tubulin antibody and DAPI to stain cellular microtubules and DNA, respectively. Immunofluorescence staining analysis of HTR8/SVneo cells by confocal microscopy showed a well-developed array of hair-like microtubule networks of slim fibrous microtubules (red) wrapped around the cell nucleus (blue) in control cells. In contrast, cells infected with
T. gondii showed a α-tubulin staining pattern that was diffuse and disorganized. In addition,
T. gondii infection induced nuclear fragmentation and cellular shrinkage, whereas untreated cells grew well with a clear complete cytoskeleton (
Fig. 1A). The numbers of
T. gondii-infected cells and the total number of cells were counted under a fluorescence microscope. As shown in
Fig. 1B, the
T. gondii infection rate significantly increased in an infection time-dependent manner. Furthermore, to evaluate the effects of
T. gondii infection on HTR8/SVneo cell viability, the cells were incubated with
T. gondii at an MOI of 10 for various times and then subjected to cell viability assay. Compared to mock-infected control cells,
T. gondii infection significantly reduced cell viability, with viabilities of 82.83±4.78%, 70.37±7.77%, and 48.50±3.49% observed for cells infected with
T. gondii for 4, 8, and 24 hr, respectively (
Fig. 1C). These results indicate that
T. gondii infection alters HTR8/SVneo cell cytoskeletal and reduces cell viability.
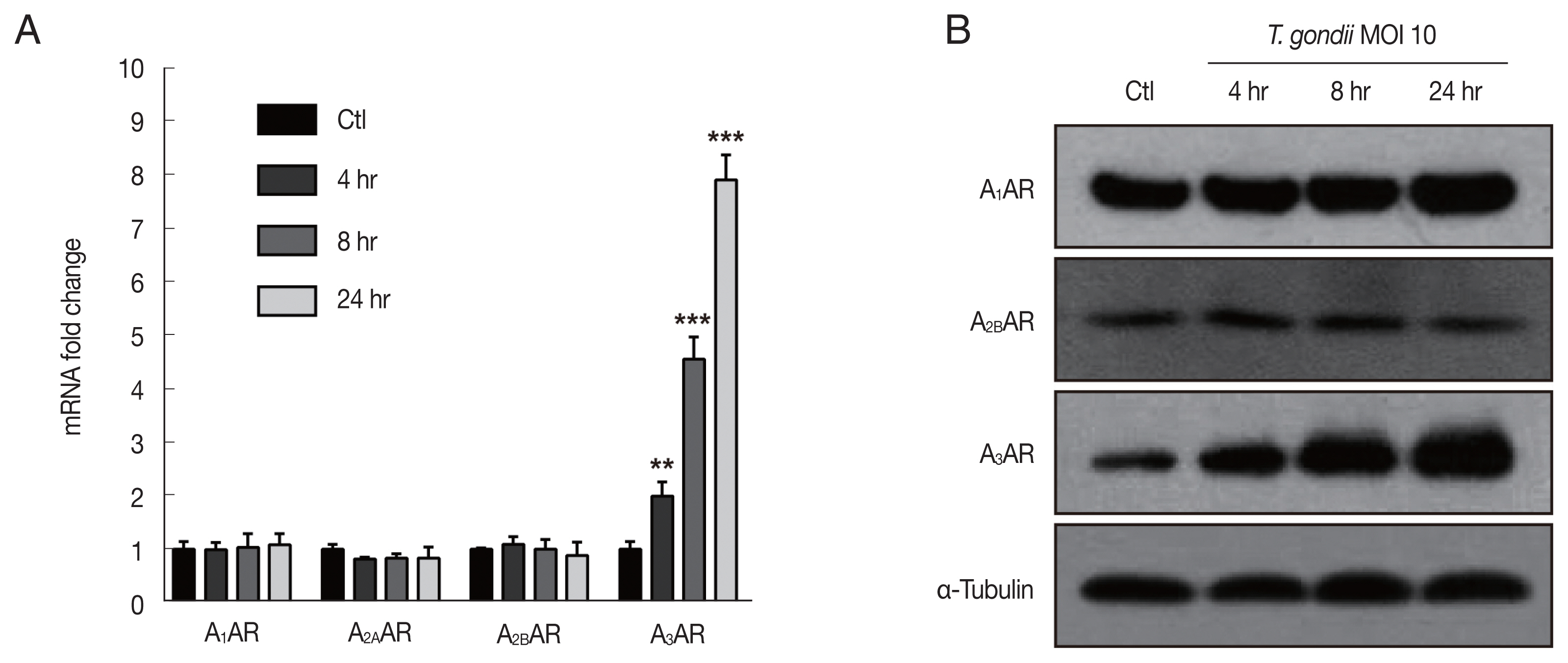
In the present study, to explore the mechanism associated with the
T. gondii-induced alterations in HTR8/SVneo cell morphology and the potential involvement of adenosine receptors, HTR8/SVneo cells were infected with
T. gondii at an MOI of 10 for 4, 8, and 24 hr. Subsequently, the gene expression of adenosine receptor family members was assessed by RT-qPCR, and the results indicated that adenosine A
3 receptor (A
3AR) mRNA levels were significantly increased after 4, 8, and 24 hr of
T. gondii infection in a time-dependent manner. Unexpectedly,
T. gondii infection did not alter A
1AR, A
2AAR, and A
2BAR mRNA expression (
Fig. 2A). We also evaluated protein levels of adenosine receptor family members by western blot analysis. Consistent with RT-qPCR results,
T. gondii infection dramatically increased A
3AR protein levels in a time-dependent manner, while those of A
1AR and A
2BAR were unaffected (
Fig. 2B). In addition, A
2AAR protein was not detected in either the control or
T. gondii-infected HTR8/SVneo cells (data not shown). These results support the hypothesis that adenosine receptors are associated with the
T. gondii-induced HTR8/SVneo cell cytoskeletal changes, which may specifically involve the time-dependent upregulation of A
3AR levels.
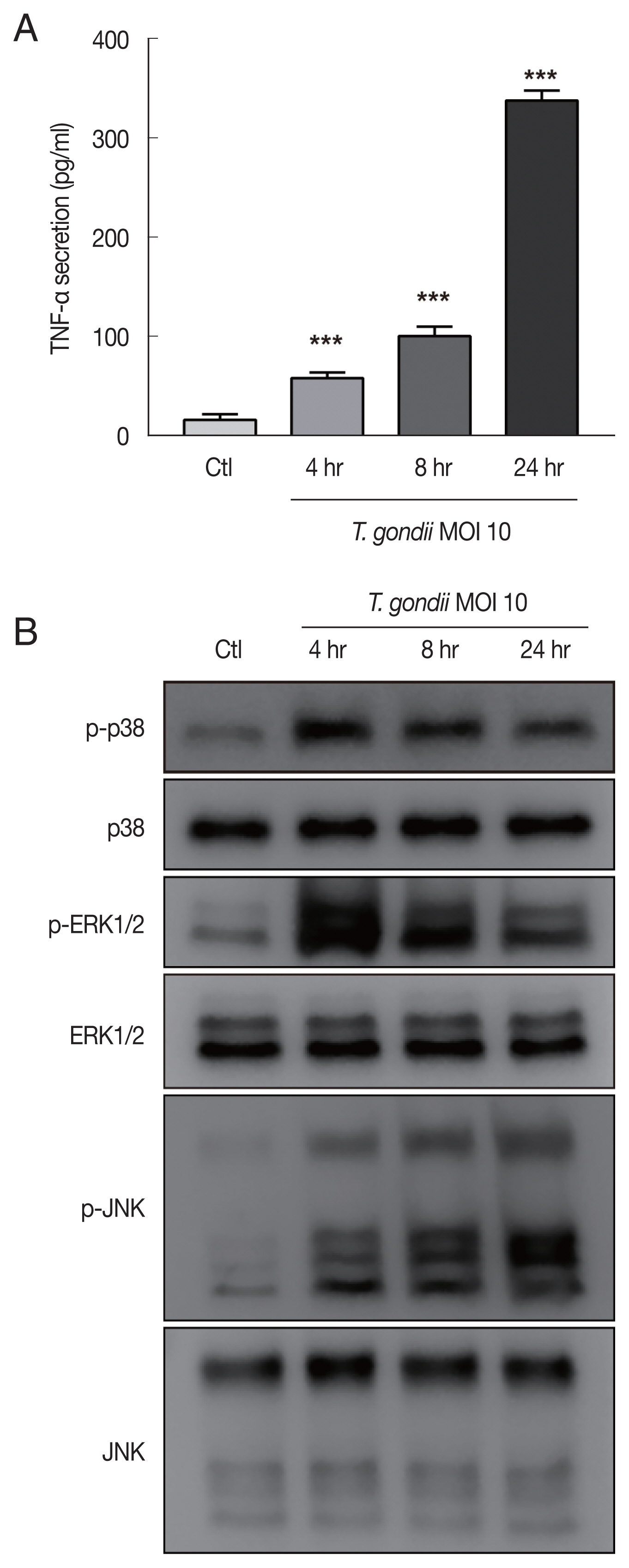
HTR8/SVneo cells were mock-infected or infected with the RH strain of
T. gondii at an MOI of 10 for 4, 8, and 24 hr and then evaluated for TNF-α secretion and MAPK activation. Significantly increased TNF-α secretion by
T. gondii-infected HTR8/SVneo cells was detected by ELISA with a time-dependent manner (
Fig. 3A). A previous study convincingly demonstrated that intracellular
T. gondii induces MAPK pathways and the production of proinflammatory cytokines in macrophages [
15]. In turn, MAPKs promote the activation of transcription factors, ultimately resulting in the production of IL-12 and TNF-α [
16], leading us to ask whether the
T. gondii-induced secretion of TNF-α by HTR8/SVneo cells was associated with the activation of MAPK. To evaluate this possibility, HTR8/SVneo cells were incubated with
T. gondii at an MOI of 10 for 4, 8, and 24 hr and then assessed for MAPK kinase pathway activation. As shown in
Fig. 3B, the phosphorylation levels of p38 and ERK1/2 protein levels were markedly increased and peaked at 4 hr postinfection and then gradually decreased. More importantly,
T. gondii infection induced the sustained phosphorylation of JNK in a time dependent manner. Based on these findings, we conclude that phosphorylation of MAPK components may be involved in the
T. gondii-induced production of TNF-α by HTR8/SVneo cells.
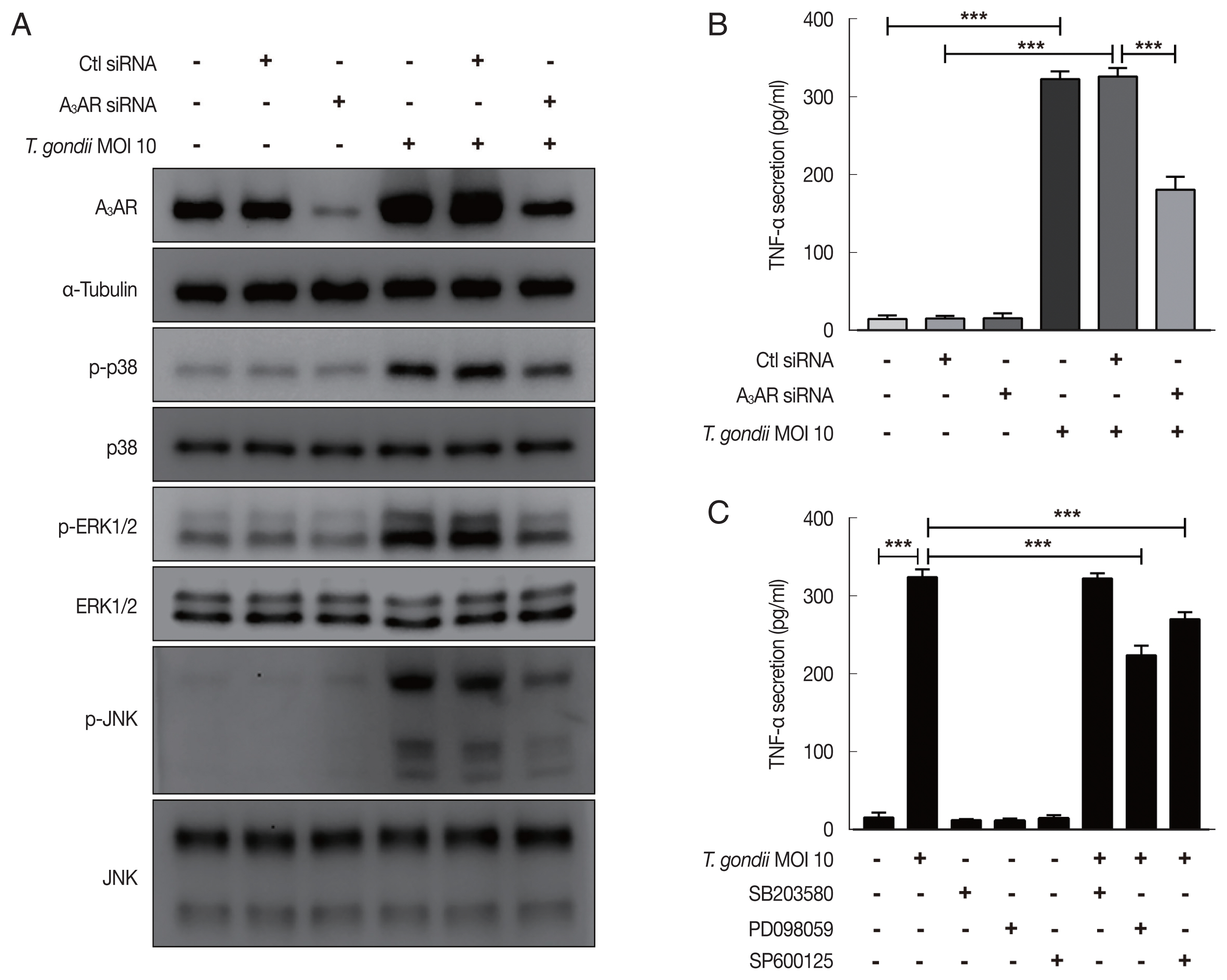
To evaluate the role of A
3AR in
T. gondii-induced MAPK activation and TNF-α production, HTR8/SVneo cells that had been incubated for 48 hr after transfection with control siRNA or A
3AR-specific siRNAs were infected with
T. gondii at an MOI of 10 for 24 hr and then MAPK activation and TNF-α secretion levels were assessed. As shown in
Fig. 4A, western blot results revealed significantly increased levels of A
3AR and phosphorylated p38, ERK1/2 and JNK in the control siRNA-transfected
T. gondii-infected cells. In contrast, the levels of these proteins were dramatically downregulated in A
3AR knockdown cells. Subsequently, to further assess whether A
3AR is involved in the regulation of TNF-α secretion, we examined TNF-α production in A
3AR siRNA-transfected
T. gondii-infected cells. Notably,
T. gondii infection markedly increased TNF-α production in control siRNA transfected cells, while A
3AR siRNA transfection significantly inhibited the
T. gondii-induced production of TNF-α (
Fig. 4B). These results strongly suggest that A
3AR is involved in the
T. gondii-induced activation of MAPK and TNF-α secretion. Subsequently, to obtain direct evidence for the association between MAPK activation and TNF-α secretion, HTR8/SVneo cells were preincubated with or without inhibitors of p38 MAPK (SB203580), ERK1/2 (PD098059), and JNK (SP600125) for 2 hr and then were infected with
T. gondii for 24 hr. ELISA results revealed that TNF-α levels were reduced by PD098059 and SP600125 (
Fig. 4C). These data indicate that A
3AR-mediated ERK1/2 and JNK signaling activation may be responsible for the increased TNF-α secretion observed in
T. gondii-infected HTR8/SVneo cells.
DISCUSSION
In the present study, we observed that T. gondii infection upregulated A3AR expression and TNF-α production in HTR8/SVneo cells in a time dependent manner. T. gondii also caused morphological changes and reduced the viability of HTR8/SVneo cells in an infection time-dependent manner. We further evaluated the effect of A3AR function by assessing the activation of MAPK signaling pathway components and the expression of TNF-α in T. gondii-infected HTR8/SVneo cells. We demonstrated that A3AR siRNA transfection downregulated TNF-α release from HTR8/SVneo cells in response to T. gondii infection and dramatically attenuated the T. gondii infection-mediated increase in p38, ERK1/2 and JNK phosphorylation levels compared to that observed in control siRNA-transfected cells. Notably, PD098059 and SP600125 pretreatment reduced the T. gondii infection mediated increase in TNF-α production. These data suggest that the effect of T. gondii infection on TNF-α release was in part regulated by A3AR-mediated ERK1/2 and JNK signaling activation.
First, we investigated whether
T. gondii infection affects HTR8/SVneo human extravillous trophoblast cell morphology and viability. We evaluated morphological changes in
T. gondii-infected HTR8/SVneo cells by assessing the integrity of the microtubule network. After infection with
T. gondii at an MOI of 10 for 4, 8, and 24 hr, we observed cells with condensed chromatin, nuclear fragmentation, and cellular shrinkage, which are associated with cell cytotoxicity [
17]. Subsequently, we further evaluated
T. gondii-induced HTR8/SVneo cell viability, and the results showed that
T. gondii infection significantly reduced cell viability, with viabilities of 82.83±4.78%, 70.37±7.77%, and 48.50±3.49% observed for cells infected with
T. gondii for 4, 8, and 48 hr, respectively. These findings are consistent with those of our previous study showing that GFP-RH (MOI 5)
T. gondii infection of human umbilical cord mesenchymal stem cells significantly reduced viability by 21 and 30% in cells infected with
T. gondii for 24 and 48 hr, respectively [
18].
Second, we investigated the mechanisms associated with the
T. gondii infection-mediated reduction in cell viability in HTR8/SVneo cells. Adenosine is a purine nucleoside involved in numerous physiological processes [
19]. Recently, Pinheiro et al. reported that adenosine A
2A receptor mediates dexamethasone induces morphological alterations in primary rat hippocampal neurons [
14]. Thus, in the present study, we evaluated whether this adenosine receptor is involved in the regulation of
T. gondii-induced HTR8/SVneo cell morphological changes.
T. gondii infection dramatically increased A
3AR mRNA and protein levels in a time-dependent manner but not those of A
1R, A
2AAR, and A
2BAR. Lima et al. previously reported that Leishmania infantum parasites subvert the host inflammatory response through adenosine A
2A receptor to promote the establishment of infection [
20]. The greatest differences between these 2 studies are the infection source of different species. In the present study, the number of
T. gondii-infected HTR8/SVneo cells and the intracellular parasite content were significantly increased in a time-dependent manner. These findings suggested that
T. gondii infection of HTR8/SVneo cells may alter morphological changes and accelerate the infection rate and intracellular proliferation through regulation of the adenosine A
3A receptor.
Third, we evaluated the functional role of A
3AR in the
T. gondii-induced production of TNF-α. Adenosine binding to A
1R and A
3R also modulates TNF-α release from adult monocytes, whereas A
2bR appears to have little effect [
21]. In the present study,
T. gondii significantly enhanced TNF-α release from HTR8/SVneo cells in a time-dependent manner, which is similar to the results of other studies [
22–
24]. However,
T. gondii exploits STAT3 to downregulate IL-12 and TNF-α expression in infected macrophages [
25]. Differences in the type of host cell,
T. gondii virulence and experimental conditions resulted in different results between these studies. A previous study convincingly demonstrated that intracellular
T. gondii induces MAPK pathway activation and the production of proinflammatory cytokines in macrophages [
15]. Regarding
T. gondii-induced intracellular signaling in HTR8/SVneo cells, Milian et al. recently showed that increased
T. gondii intracellular proliferation in human extravillous trophoblast cells (HTR8/SVneo line) is sequentially triggered by MIF, ERK1/2, and COX-2 [
2]. Thus, we further evaluated
T. gondii-induced MAPK intracellular signaling pathway activation in HTR8/SVneo cells.
T. gondii dramatically increased the phosphorylation levels of p38 and ERK1/2, which peaked at 4 hr postinfection and then gradually decreased. In addition,
T. gondii induced sustained phosphorylation of JNK in time-dependent manner. Subsequently, the role of A
3AR in
T. gondii-induced MAPK activation and TNF-α production were examined.
T. gondii significantly increased TNF-α production, A
3AR protein levels and the phosphorylation levels of p38, ERK1/2 and JNK in control siRNA-transfected
T. gondii-infected cells. Moreover, the inhibition of A
3AR by A
3AR siRNA transfection apparently suppressed the
T. gondii-mediated upregulation of TNF-α and A
3AR levels and the activation of MAPK. These results are supported by those of many other studies showing that adenosine receptors are involved in MAPK activation and TNF-α secretion [
9,
21]. Finally, the role of MAPK activation in the
T. gondii-mediated regulation of TNF-α secretion was evaluated by TNF-α ELISA. The results showed that the
T. gondii-mediated increase in TNF-α secretion was dramatically attenuated by pretreatment of cells with PD098059 or SP600125. These results indicate that the A
3AR-mediated activation of ERK1/2 and JNK positively regulate TNF-α secretion in
T. gondii-infected HTR8/SVneo cells.
In summary, in the present study, we elucidated a mechanism of adenosine A3 receptor-mediated ERK1/2- and JNK-dependent TNF-α production in T. gondii-infected HTR8/SVneo human extravillous trophoblast cells that ultimately may induce abnormal pregnancy. This is the first study to systemically evaluate the effect of T. gondii infection on the adenosine receptor family proteins and the regulatory mechanism of TNF-α production mediated by adenosine A3 receptor. Further studies are need to assess the role of T. gondii infection in placental development and possibly in the pathophysiology of preeclampsia.
Notes
-
CONFLICT OF INTREST
The authors declare that they have no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81771612), the Science Foundation of Guangdong Medical University (GDMUZ2019003), the Characteristic Innovation Projects of Guangdong Universities (2018KTSCX081), and the Natural Science Foundation of Guangdong Province (2019A1515011715).
Fig. 1
Toxoplasma gondii infection induced morphological changes in HTR8/SVneo cells and reduces cell viability. (A) HTR8/SVneo cells were infected with GFP-expressing T. gondii at an MOI of 10 for the indicated time durations. Cells were fixed and probed against α-tubulin (red), after which they were counterstained with DAPI (blue) and visualized by confocal microscopy. Scale bar=30 μm. (B) Number of T. gondii-infected cells and total number of cells were counted to calculate infection rate. (C) Cell viability was measured by using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit (mean±SD). *P<0.05, **P<0.01, ***P<0.001 compared with mock-infected control cells.

Fig. 2
Toxoplasma gondii infection upregulated adenosine A3 receptor expression in HTR8/SVneo cells. HTR8/SVneo cells were infected with the RH strain of T. gondii at an MOI of 10 for the indicated time durations. (A) Adenosine A1, A2A, A2B and A3 receptor expression assessed by RT-qPCR. HPRT1 was used as an internal control. **P<0.01, ***P<0.001 compared with mock-infected control cells. (B) Expression level of adenosine receptors assessed by western blot analysis. α-Tubulin used as the loading control.

Fig. 3
Toxoplasma gondii infection increased TNF-α secretion and MAPK activation in HTR8/SVneo cells. HTR8/SVneo cells were infected with T. gondii at an MOI of 10 for the indicated time durations. (A) TNF-α secretion levels evaluated by ELISA. ***P<0.001 compared with mock-infected control cells. (B) Expression level of MAPK pathway molecules assessed by western blots analysis. α-Tubulin was used as a loading control.

Fig. 4Roles of A3AR in T. gondii-induced MAPK activation and TNF-α secretion in HTR8/SVneo cells. HTR8/SVneo cells were transfected with control siRNA or A3AR-specific siRNAs and infected with T. gondii at an MOI of 10 for 24 hr. (A) Expression level of A3AR and MAPK pathway molecules assessed by western cell culture supernatants blot α-Tubulin was used as a loading control. (B) Concentration of TNF-α in was evaluated by ELISA. (C) Inhibitory effect of p38 MAPK (SB203580), ERK1/2 (PD098059), and JNK (SP600125) on TNF-α secretion levels in T. gondii-infected cells. HTR8/SVneo cells were pretreated with or without 30 μM SB203580, PD098059 or SP600125 for 2 hr and subsequently infected with T. gondii at an MOI 10 of for 24 hr. TNF-α secretion levels was evaluated by ELISA. ***P<0.001 compared with respective control.

Table 1Adenosine receptor family primer sequences used for quantitative reverse transcription PCR (RT-qPCR) in the present study
Table 1
|
Gene name |
GenBank Accession No. |
Primer sequence (5′-3′) |
Product size (bp) |
|
ADORA1 (A1AR) |
NM_000674.3 |
F-ATTGCTGTGGACCGCTACCTCC |
153 |
|
|
R-CGCACTCAGATTGTTCCAGCCA |
|
|
|
ADORA2 (A2AAR) |
NM_000675.6 |
F-ACCGCTACATTGCCATCCGCAT |
151 |
|
|
R-TCCTTTGGCTGACCGCAGTTGT |
|
|
|
ADORA2B (A2BAR) |
NM_000676.2 |
F-GCTCCATCTTCAGCCTTCTGGC |
125 |
|
|
R-AAGGACCCAGAGGACAGCAATG |
|
|
|
ADORA3 (A3AR) |
BC029831.1 |
F-ATACAAGAGGGTCACCACTCA |
204 |
|
|
R-CAGGTGAGGAAGCTGAAGTATAC |
|
|
|
HPRT1 |
NM_000194.2 |
F-GACCAGTCAACAGGGGACAT |
111 |
|
|
R-CTGCATTGTTTTGCCAGTGT |
|
References
- 1. Flegr J, Prandota J, Sovičková M, Israili ZH. Toxoplasmosis--a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS One 2014;9:e90203.
- 2. Milian ICB, Silva RJ, Manzan-Martins C, Barbosa BF, Guirelli PM, Ribeiro M, de Oliveira Gomes A, Ietta F, Mineo JR, Silva Franco P, Ferro EAV. Increased Toxoplasma gondii intracellular proliferation in human extravillous trophoblast cells (HTR8/SVneo Line) is sequentially triggered by MIF, ERK1/2, and COX-2. Front Microbiol 2019;10:852.
- 3. Barbosa BF, Silva DA, Costa IN, Mineo JR, Ferro EA. BeWo trophoblast cell susceptibility to Toxoplasma gondii is increased by interferon-gamma, interleukin-10 and transforming growth factor-beta1. Clin Exp Immunol 2008;151:536-545.
- 4. Nishikawa Y, Kawase O, Vielemeyer O, Suzuki H, Joiner K, Xuan X, Nagasawa H.
Toxoplasma gondii infection induces apoptosis in noninfected macrophages: role of nitric oxide and other soluble factors. Parasite Immunol 2007;29:375-385.
- 5. Abbasi M, Kowalewska-Grochowska K, Bahar MA, Kilani RT, Winkler-Lowen B, Guilbert LJ. Infection of placental trophoblasts by Toxoplasma gondii
. J Infect Dis 2003;188:608-616.
- 6. Suwanti LT, Mufasirin M. Peningkatan TNF-α dan indeks apoptosis pada tulang mencit yang diinfeksi Toxoplasma gondii
. J Ked Hewan 2015;9:101-104. (in Suroboyoan)..
- 7. Toder V, Fein A, Carp H, Torchinsky A. TNF-α in pregnancy loss and embryo maldevelopment: a mediator of detrimental stimuli or a protector of the fetoplacental unit? J Assist Reprod Genet 2003;20:73-81.
- 8. Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov 2008;7:759-770.
- 9. Darashchonak N, Sarisin A, Kleppa MJ, Powers RW, von Versen-Hoynck F. Activation of adenosine A2B receptor impairs properties of trophoblast cells and involves mitogen-activated protein (MAP) kinase signaling. Placenta 2014;35:763-771.
- 10. Borroto-Escuela DO, Hinz S, Navarro G, Franco R, Müller CE, Fuxe K. Understanding the role of adenosine A2AR heteroreceptor complexes in neurodegeneration and neuroinflammation. Front Neurosci 2018;12:43.
- 11. Iriyama T, Xia Y. Placental Adenosine Signaling in the Pathophysiology of Preeclampsia. In Saito S ed, Preeclampsia. Singapore. Springer. 2018, pp 99-112.
- 12. Ko HS, Choi SK, Kang HK, Kim HS, Jeon JH, Park IY, Shin JC. Oncostatin M stimulates cell migration and proliferation by down-regulating E-cadherin in HTR8/SVneo cell line through STAT3 activation. Reprod Biol Endocrinol 2013;11:93.
- 13. Liu Y, Shan N, Yuan Y, Tan B, He C, Tong C, Qi H. Knockdown of activated Cdc42-associated kinase inhibits human extravillous trophoblast migration and invasion and decreases protein expression of pho-Akt and matrix metalloproteinase. J Matern Fetal Neonatal Med 2020;33:1125-1133.
- 14. Pinheiro H, Gaspar R, Baptista FI, Fontes-Ribeiro CA, Ambrósio AF, Gomes CA. Adenosine A2A receptor blockade modulates glucocorticoid-induced morphological alterations in axons, but not in dendrites, of hippocampal neurons. Front Pharmacol 2018;9:219.
- 15. Quan JH, Chu JQ, Kwon J, Choi IW, Ismail HA, Zhou W, Cha GH, Zhou Y, Yuk JM, Jo EK, Lee YH. Intracellular networks of the PI3K/AKT and MAPK pathways for regulating Toxoplasma gondii-induced IL-23 and IL-12 production in human THP-1 cells. PLoS One 2015;10:e0141550.
- 16. Denkers EY, Butcher BA, Del Rio L, Kim L. Manipulation of mitogen-activated protein kinase/nuclear factor-κB-signaling cascades during intracellular Toxoplasma gondii infection. Immunol Rev 2004;201:191-205.
- 17. Quan JH, Gao FF, Ismail HAHA, Yuk JM, Cha GH, Chu JQ, Lee YH. Silver nanoparticle-induced apoptosis in ARPE-19 cells is inhibited by Toxoplasma gondii pre-infection through suppression of NOX4-dependent ROS generation. Int J Nanomedicine 2020;15:3695-3716.
- 18. Chu JQ, Jing KP, Gao X, Li P, Huang R, Niu YR, Yan SQ, Kong JC, Yu CY, Shi G, Fan YM, Lee YH, Zhou Y, Quan JH.
Toxoplasma gondii induces autophagy and apoptosis in human umbilical cord mesenchymal stem cells via downregulation of Mcl-1. Cell Cycle 2017;16:477-486.
- 19. Feoktistov I, Biaggioni I. Adenosine A2B receptors. Pharmacol Rev 1997;49:381-402.
- 20. Lima MH, Sacramento LA, Quirino GF, Ferreira MD, Benevides L, Santana AK, Cunha FQ, Almeida RP, Silva JS, Carregaro V.
Leishmania infantum parasites subvert the host inflammatory response through the adenosine A2A receptor to promote the establishment of infection. Front Immunol 2017;8:815.
- 21. Chavez-Valdez R, Wills-Karp M, Ahlawat R, Cristofalo EA, Nathan A, Gauda EB. Caffeine modulates TNF-α production by cord blood monocytes: the role of adenosine receptors. Pediatr Res 2009;65:203.
- 22. Angeloni MB, Guirelli PM, Franco PS, Barbosa BF, Gomes AO, Castro AS, Silva NM, Martins-Filho OA, Mineo TWP, Silva DAO, Mineo JR, Ferro EAV. Differential apoptosis in BeWo cells after infection with highly (RH) or moderately (ME49) virulent strains of Toxoplasma gondii is related to the cytokine profile secreted, the death receptor Fas expression and phosphorylated ERK1/2 expression. Placenta 2013;34:973-982.
- 23. Li Y, Xiu F, Mou Z, Xue Z, Du H, Zhou C, Li Y, Shi Y, He S, Zhou H. Exosomes derived from Toxoplasma gondii stimulate an inflammatory response through JNK signaling pathway. Nanomedicine 2018;13:1157-1168.
- 24. Belloni A, Aubert D, Marin JG, Le Naour R, Bonhomme A, Guenounou M, Pinon J. Involvement of tumor necrosis factor-α during infection of human monocytic cells by Toxoplasma gondii
. Parasitol Res 2000;86:406-412.
- 25. Butcher BA, Kim L, Panopoulos AD, Watowich SS, Murray PJ, Denkers EY. Cutting edge: IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-α in host macrophages. J Immunol 2005;174:3148-3152.

 , Jinhui Sun2,†
, Jinhui Sun2,†