Abstract
The first Korean case of Echinococcus multilocularis interpreted the probable origin of the isolate on the basis of cox1 sequence similarity, originally reporting 99.8%–99.9% similarity to Asian isolates and 99.5%–99.6% similarity to European isolates. To examine whether this similarity-based interpretation remains epidemiologically informative, we re-evaluated currently available cox1 sequences with high sequence identity at the accession level. Using AB780998.1 as the query, BLAST hits with ≥99.5% identity were collected, and 110 non-self accessions were manually annotated according to clade classifications reported in the source literature. Sequence alignment and phylogenetic assessment were performed with MAFFT and IQ-TREE. Across the re-evaluated dataset, sequences with identities from 99.5025% to 99.6269% were predominantly classified as European, whereas all sequences from 99.6891% to 99.9378% were classified as Asian. The only 2 exceptions to this overall pattern were OR263180 and OR263183, which were classified as Asian despite falling within the lower identity band. These 2 accessions were reported in a study using a concatenated mitochondrial framework including cox1, cob, and nad2, suggesting that single-marker interpretation may be limited in such borderline cases. In the 1,608 bp alignment, 1,544 sites were constant and only 19 were parsimony-informative. Despite this limited variation, cox1 percent identity showed strong concordance with classification of Asian and European groups reported in the source literature. These findings indicate that high cox1 identity may serve as a practical proxy for broad lineage discrimination between Asian and European groups of E. multilocularis, although rare borderline cases may require confirmation with multiple mitochondrial markers.
-
Key words: Echinococcus multilocularis, alveolar echinococcosis, mitochondrial DNA, sequence analysis, phylogeny
Echinococcus multilocularis is the causative agent of alveolar echinococcosis, a potentially fatal zoonotic disease in humans, and is distributed predominantly throughout the Northern Hemisphere [
1]. A previous mitochondrial phylogeographic study has identified the broad geographic structuring of
E. multilocularis corresponding to European, Asian, and North American clades [
2]. In Korea, where indigenous infection had not previously been documented, the first reported case of
E. multilocularis was interpreted epidemiologically on the basis of cytochrome c oxidase subunit 1 (
cox1) sequence similarity [
3]. The present study re-examines whether this similarity-based interpretation remains epidemiologically valid in light of currently available accession-level sequence data.
The original Korean report demonstrated that the Korean isolate shared 99.8%–99.9%
cox1 sequence similarity with Asian isolates, with the highest similarity to an isolate from Sichuan, China, whereas similarity with European isolates ranged from 99.5% to 99.6% [
3]. In this regional context,
E. multilocularis populations from the Western Sichuan Plateau have been reported to exhibit low genetic variation based on microsatellite and mitochondrial markers, with partial
cox1 haplotypes assigned to the Asian clade [
4]. Although the original interpretation has been regarded as epidemiologically informative, its validity at the accession level has not been reassessed against the larger number of highly similar
cox1 accessions currently available in public databases. Accordingly, we revisited this issue by evaluating whether the identity ranges described in the original Korean case remain concordant with Asian and European classifications in the source literature when currently retrievable accessions with high sequence identity are examined individually. Therefore, the present study focused on an accession-level re-evaluation of the epidemiologic interpretation of the first Korean case, rather than proposing a revised phylogeographic framework for
E. multilocularis.
Using AB780998.1 as the query sequence, BLAST hits with ≥99.5% identity were collected. After excluding the query sequence itself, 110 accessions remained for evaluation. This study was intentionally restricted to the high-identity interval relevant to the original Korean report and was not designed to assess the full sequence diversity of
E. multilocularis. For each accession, geographic metadata were compiled, and the corresponding source study was manually identified. Lineage assignments were adopted from the corresponding source literature and were not inferred de novo from geographic origin or phylogenetic reconstruction in the present study. The lineage terminology used here follows the continental-scale grouping framework for
E. multilocularis originally described by Nakao et al. [
2]. Complete accession-level annotations are presented in
Supplementary Table S1. Geographic metadata (geo_loc_name) were obtained from NCBI, whereas lineage classification was based on a manual review of the corresponding source studies listed in
Supplementary Table S1.
As a complementary analysis, the 110
cox1 sequences were aligned using MAFFT [
5] and analyzed in IQ-TREE 3 by ModelFinder and ultrafast bootstrap analysis [
6-
8]. In the resulting alignment comprising 1,608 bp, 1,544 sites were constant, and only 19 sites were parsimony-informative. ModelFinder identified HKY+F+G4 as the best-fit model according to the Bayesian information criterion [
6,
7]. The small number of informative sites indicated limited phylogenetic resolution from this marker alone, particularly for finer internal relationships [
9]. Consistent with this observation, bootstrap support across internal nodes was generally low (median, 17; mean, 32.77; range, 1–99). Nevertheless, despite the weak internal support caused by high sequence homogeneity, the distribution of percent identity values showed clear separation between the 2 broad geographical groups.
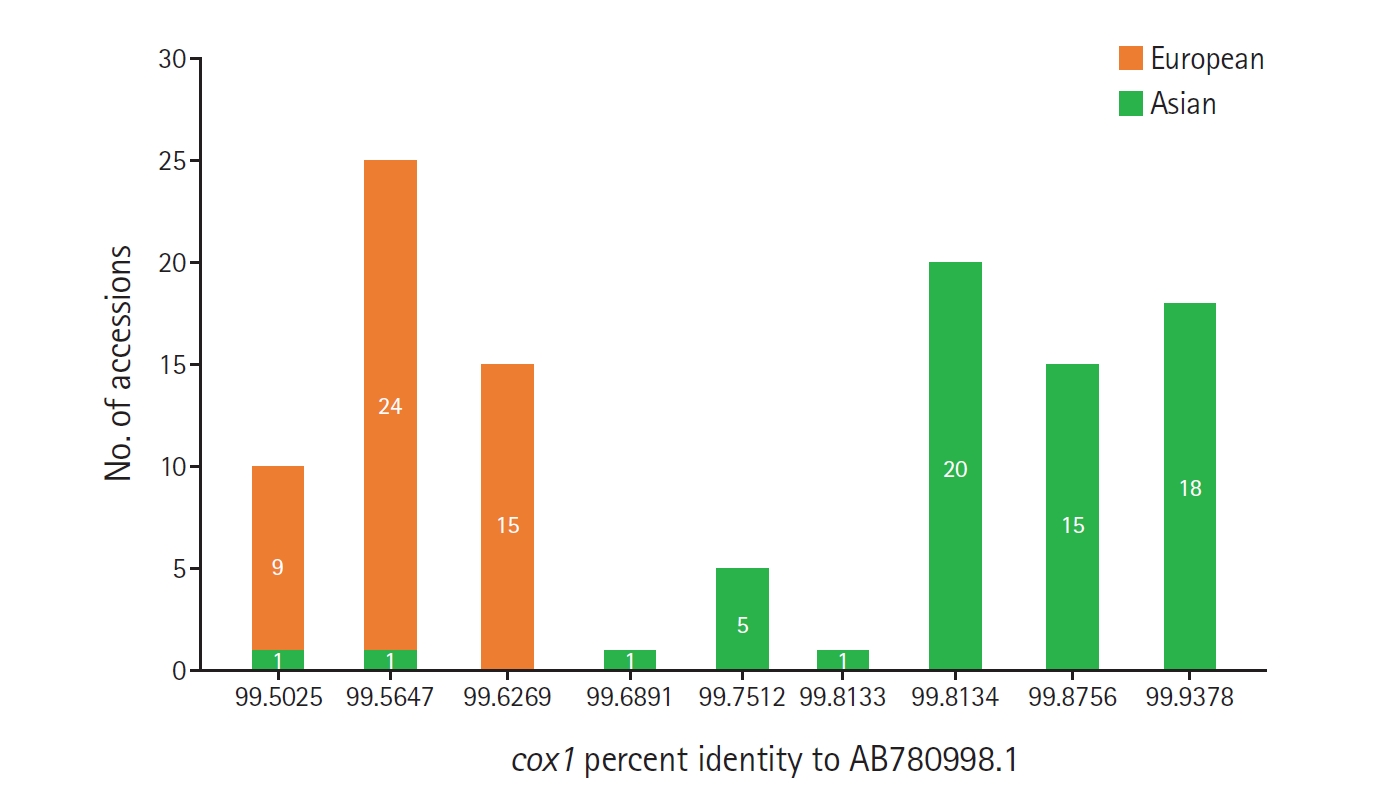
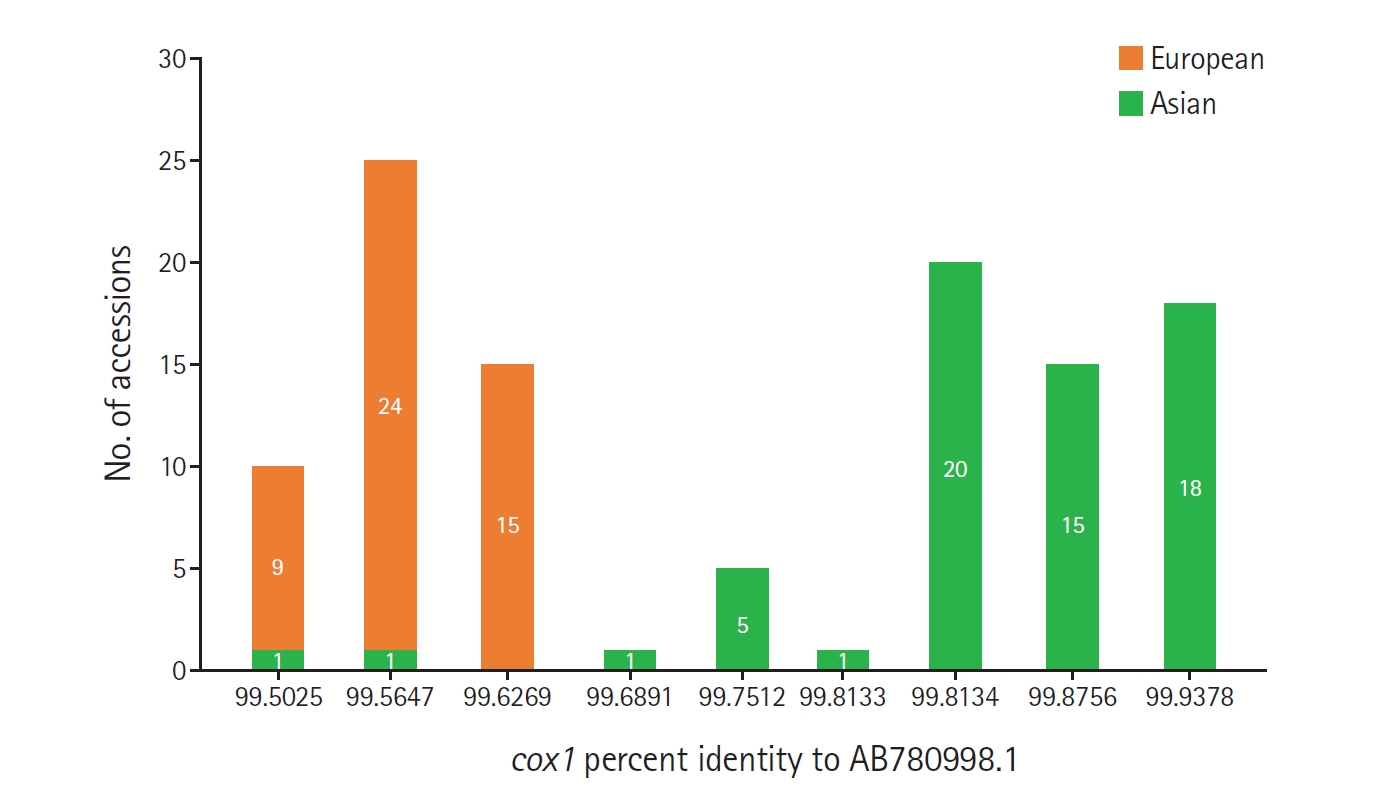
Despite the limited phylogenetic signal, a distinct pattern of segregation by percent identity was observed. Accessions with percent identities ranging from 99.5025% to 99.6269% were predominantly classified as European in the source literature, with only 2 Asian-classified exceptions (OR263180 and OR263183). In contrast, all accessions with identities ranging from 99.6891% to 99.9378% were classified as Asian. Therefore, within the ≥99.5% identity interval, lower identity values corresponded predominantly to European-classified sequences, whereas higher identity values were exclusively associated with Asian-classified sequences (
Fig. 1). These findings support the epidemiologic interpretation proposed in the original Korean report [
3].
The 2 exceptional accessions (OR263180 and OR263183) were both reported in a study conducted in Yili Prefecture, Xinjiang [
10]. In that study, lineage interpretation was based on a concatenated mitochondrial framework incorporating
cox1, cytochrome b (
cob), and NADH dehydrogenase subunit 2 (
nad2); notably, both accessions were linked to the same
cob accession (OR243690) rather than being evaluated on the basis of
cox1 alone [
10]. Therefore, these cases may be more appropriately regarded as boundary cases illustrating the limitations of single-marker interpretation rather than as direct contradictions to the overall segregation pattern. The purpose of identifying these exceptions is not to resolve their phylogeographic origin but to demonstrate that
cox1 identity can still provide a practical epidemiologic proxy under conditions of limited sequence information. However, in borderline or discordant cases, lineage interpretation should be approached cautiously and supported by additional mitochondrial markers (e.g.,
cob and
nad2) or by previously established multi-marker classification frameworks.
Even within this highly homogeneous dataset, percent identity retained substantial classificatory signal. Beyond simply expanding the comparison set, the present study shows that even within a narrow high-identity interval,
cox1 percent identity in
E. multilocularis remained informative at a broad phylogeographic level, with lower identity values corresponding predominantly to European-classified accessions and higher identity values confined to Asian-classified accessions. This observation is notable because, despite the limited number of parsimony-informative sites and generally low internal bootstrap support, these identity ranges remained strongly concordant with source-based continental classification. Although variation across the 1,608 bp region was limited, identity values remained strongly associated with annotation as Asian or European in the source literature. Taken together, these findings suggest that the
cox1 similarity ranges reported for the first Korean case were not merely descriptive but were broadly consistent with later evidence evaluated at the accession level [
3]. The present analysis does not argue that percent identity should replace phylogenetic or haplotype-based inference in general. Rather, it shows that in the present
E. multilocularis dataset,
cox1 percent identity was strongly concordant with source-based classification of Asian and European groups, allowing percent identity to function as a practical epidemiologic proxy, while rare borderline cases may benefit from confirmation with additional mitochondrial markers such as
cob or
nad2 [
10].
Notes
-
Author contributions
Conceptualization: Kim J. Data curation: Kim J. Formal analysis: Kim J. Investigation: Kim J. Methodology: Kim J. Supervision: Kwak D. Validation: Kim J, Kwak D. Visualization: Kim J. Writing-original draft: Kim J. Writing-review & editing: Kim J, Kwak D.
-
Conflict of interest
Dongmi Kwak serves as an editor of Parasites, Hosts and Diseases but had no involvement in the decision to publish this article. No other potential conflicts of interest relevant to this study were reported.
Supplementary information
Fig. 1.Frequency distribution of cox1 percent identity values relative to AB780998.1 among 110 non-self Echinococcus multilocularis accessions classified as Asian or European in the source literature. Bars indicate the number of accessions assigned to each group at each observed percent identity value.
References
- 1. Baumann S, Shi R, Liu W, et al. Worldwide literature on epidemiology of human alveolar echinococcosis: a systematic review of research published in the twenty-first century. Infection 2019;47:703-27. https://doi.org/10.1007/s15010-019-01325-2
- 2. Nakao M, Xiao N, Okamoto M, et al. Geographic pattern of genetic variation in the fox tapeworm Echinococcus multilocularis. Parasitol Int 2009;58:384-9. https://doi.org/10.1016/j.parint.2009.07.010
- 3. Jeong JS, Han SY, Kim YH, et al. Serological and molecular characteristics of the first Korean case of Echinococcus multilocularis. Korean J Parasitol 2013;51:595-7. https://doi.org/10.3347/kjp.2013.51.5.595
- 4. Shang JY, Zhang GJ, Liao S, et al. Low genetic variation in Echinococcus multilocularis from the Western Sichuan Plateau of China revealed by microsatellite and mitochondrial DNA markers. Acta Trop 2021;221:105989. https://doi.org/10.1016/j.actatropica.2021.105989
- 5. Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002;30:3059-66. https://doi.org/10.1093/nar/gkf436
- 6. Wong TKF, Ly-Trong N, Ren H, et al. IQ-TREE 3: phylogenomic inference software using complex evolutionary models [Preprint]. Posted 2025 Apr 7. EcoEvoRxiv. https://doi.org/10.32942/X2P62N
- 7. Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 2017;14:587-9. https://doi.org/10.1038/nmeth.4285
- 8. Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 2018;35:518-22. https://doi.org/10.1093/molbev/msx281
- 9. Townsend JP, Su Z, Tekle YI. Phylogenetic signal and noise: predicting the power of a data set to resolve phylogeny. Syst Biol 2012;61:835-49. https://doi.org/10.1093/sysbio/sys036
- 10. Guo B, Cairen , Wu J, et al. The A2 haplotype of Echinococcus multilocularis is the predominant variant infecting humans and dogs in Yili Prefecture, Xinjiang. Infect Genet Evol 2024;119:105581. https://doi.org/10.1016/j.meegid.2024.105581
Citations
Citations to this article as recorded by