Abstract
Toxoplasma gondii provokes rapid and sustained nuclear translocation of the signal transducer and activator of transcription 6 (STAT6) in HeLa cells. We observed activation of STAT6 as early as 2 hr after infection with T. gondii by the nuclear translocation of fluorescence expressed from exogenously transfected pDsRed2-STAT6 plasmid and by the detection of phosphotyrosine-STAT6 in Western blot. STAT6 activation occurred only by infection with live tachyzoites but not by co-culture with killed tachyzoites or soluble T. gondii extracts. STAT6 phosphorylation was inhibited by small interfering RNA of STAT6 (siSTAT6). In view of the fact that STAT6 is a central mediator of IL-4 induced gene expression, activation of STAT6 by T. gondii infection resembles that infected host cells has been stimulated by IL-4 treatment. STAT1 was affected to increase the transcription and expression by the treatment of siSTAT6. STAT6 activation was not affected by any excess SOCS's whereas that with IL-4 was inhibited by SOCS-1 and SOCS-3. T. gondii infection induced Eotaxin-3 gene expression which was reduced by IFN-γ. These results demonstrate that T. gondii exploits host STAT6 to take away various harmful reactions by IFN-γ. This shows, for the first time, IL-4-like action by T. gondii infection modulates microbicidal action by IFN-γ in infected cells.
-
Key words: Toxoplasma gondii, entry, STAT6 phosphorylation, nuclear translocation, STAT1, IL-4, IFN-γ
INTRODUCTION
Toxoplasma gondii is an obligate intracellular protozoan parasite and an important zoonotic pathogen that causes severe diseases in congenitally infected patients and in immunocompromised patients such as AIDS victims in addition to the parasite infecting healthy persons [
1].
T. gondii infects macrophages primarily and is capable of invading and replicating within a wide variety of nucleated host cells.
The signal transducer and activator of transcription (STAT) family of transcription factors mediate various extracellular cytokine signals into diverse biological responses via the modulation of gene transcription specific to the cytokines [
2]. The participation of STAT proteins in immune regulation by cytokines from
T. gondii infected host cells has been well established [
3]. It has been well known that macrophages, dendritic cells, and polymorphonuclear cells secrete IL-12 in response to
T. gondii infection [
4,
5]. IL-12, acting through STAT4, promotes the production of IFN-γ. Through intracellular signaling intermediates such as STAT1 and T-bet, IFN-γ can induce differentiation of Th1 lymphocytes and possibly CD8
+ and NK cells [
6]. IFN-γ signaling through STAT1 is also involved in promoting IL-12 production by antigen-presenting cells. More recent evidence indicates that STAT4 signaling promotes IFN-γ production from dendritic cells and macrophages. The major antimicrobial effects of IFN-γ during
T. gondii infection, the generation of nitric oxide and induction of IFN-γ inducible GTP binding protein (IGTP) and LRG-47, are mediated through STAT1 [
7,
8]. Recently, it has been shown that cell invasion by
T. gondii activates STAT3 of macrophages, which suppresses LPS-triggered TNF-α and IL-12 production [
9].
We present, in this study, the evidence of nuclear translocation of STAT6 in the signaling during infection with T. gondii in host cells. This activation of host STAT6 after intracellular penetration of T. gondii resembles that of host cell stimulation by IL-4, which protects against the toxoplasmacidal action of IFN-γ.
MATERIALS AND METHODS
Parasite and host cells
The RH strain of
T. gondii was maintained by peritoneal passage in BALB/c mice. Prior to use, the tachyzoites were purified by centrifugation over 40% Percoll (Amersham Parmacia Biotech, Uppsala, Sweden) in PBS solution [
10]. HeLa (ATCC CCL-2) cells were cultured in minimum essential medium (MEM) supplemented with 10% FBS and used as host cells.
Total RNA was isolated from HeLa cells using TRIzol reagent (Invitrogen, Carlsbad, California, USA) according to the instruction provided by the manufacturer. The first strand cDNA was synthesized from 1 µg of total RNA by using the Powerscript reverse transcriptase (BD Biosciences Clontech, Mountain View, California, USA). For the STAT1, PCR was performed with a forward primer of 5'-GAG CTC ATG TCT CAG TGG TAC GAC-3' and reverse primer of 5'-CGG TAC CGT GTT CAT CAT ACT GTC-3' to amplify 2,253 bp STAT1α gene. The DNA fragments were inserted into green fluorescent plasmid pEGFP-N1 expression vector (BD Biosciences Clontech). For STAT6, a forward primer 5'-ACC CTC GAG TCC GCC ACC ATG GCT CTG TGG GGT CTG-3' and reverse primer 5'-CAG CTG GGA TCG AAT TCT GGG GTT GGC CCT-3' were used to amplify 2,570 bp to insert into red fluorescent plasmid pDsRed2-N1 expression vector (BD Biosciences Clontech).
Transient transfection of HeLa cells
Transient transfection of HeLa cells was achieved using calcium phosphate coprecipitation [
11]. The day before transfection, 5 × 10
4 cells were seeded into 24-well culture plates in fresh medium. Plasmid DNA (1-2 µg) was diluted in 42 µl of H
2O, mixed with 7 µl of 2 M CaCl
2 and added by drops to 50 µl of 2 × HeBS (280 mM NaCl, 1.5 mM Na
2HPO
4, and 50 mM HEPES, pH 7.05). After a 20-min incubation at room temperature, the mixture was added to the cells. After 24 hr, cells were washed and cultured for 24 hr in the presence or absence of 100 ng/ml IL-4 (Cytolab Ltd, Rehovot, Israel) or IFN-γ (Upstate, Charlottesville, Virginia, USA) were infected with RH tachyzoites of 1 × 10
8 per ml. Cells were fixed in 4% paraformaldehyde-PBS for 10 min. Hoechst dye 33258 (Invitrogen) was added to stain cell nuclei before mounting with coverslips. Fluorescence was observed under a fluorescence microscopy (Axiophot, Carl Zeiss, Oberkochen, Germany).
The proteins were resolved in 12% SDS-PAGE and transferred onto nitrocellulose (NC) sheets [
12]. The NC sheets blocked with 5% skim milk in PBS/0.05% Tween-20 were incubated with 1 : 1,000 diluted rabbit anti-human STAT1α, phosphotyrosine-STAT1α, STAT6, or phosphotyrosine-STAT6 antibody (Cell Signaling Technology, Danvers, Massachusetts, USA), and then with 1 : 5,000 diluted HRP-conjugated goat anti-rabbit IgG antibody (Santa Cruz Biotechnology, Santa Cruz, California, USA). They were soaked in enhanced chemiluminescence (ECL) solution (Amersham Pharmacia Biotech) for 1 min and then exposed to an x-ray film (Konica, Tokyo, Japan).
STAT6 activation was shut down directly by interference with transcribed STAT6 mRNA by 5'-AAG CAG GAA GAA CUC AAG UUU TT-3' and 5'-AAA CUU GAG UUC UUC CUG CUU TT-3' [
13]. Cells cultured on coverslips in a 24-well plate were transfected with a premix of 6 pmole siRNA and Lipofectamin RNAi MAX (Invitrogen) in Opti-MEM for 6 hr. After washing with MEM, cells were further incubated for 24 hr.
RT-PCR was performed with a forward primer of 5'-TTC CTC GAG ATG GTA GCA CAG AAC CAG GTG-3' and reverse primer of 5'-TGC AAG CTT AAT CTG GAA GGG GAA GGA GC-3' for SOCS-1 (651 bp), forward primer of 5'-TGC CTC GAG GAC CCT TCT CTC ATG ACC-3' and reverse primer of 5'-AGT GGA TCC GGT AAA GGC AGT CCC CA-3' for SOCS-2 (581 bp), and forward primer of 5??CCG CTC GAG ATG GTC ACC CAC AGC-3' and reverse primer of 5'-TAC AAG CTT AAG CGG GGC ATC GTA CTG GTC-3' for SOCS-3 (693 bp), respectively. Amplified DNA fragments were inserted into the pEGFP-N1 expression vector (BD Biosciences Clontech). After transfection with SOCS-1, -2, and -3, cells were treated with IL-4 or infected with T. gondii. Cells were lysed and blotted on the phosphotyrosine-STAT6 and STAT6.
Quantitative RT-PCR and promoter assay of Eotaxin-3 (CCL26)
The RT-PCR product of Eotaxin-3 was measured quantitatively with the combination of IFN-γ, IL-4, and
T. gondii infection [
11]. The forward primer of 5'-GGA ACT GCC ACA CGT GGG AGT GAC-3' and reverse primer of 5??CTC TGG GAG GAA ACA CCC TCT CC-3' were used to amplify 354 bp CCL26 DNA with the treatment of IFN-γ, IL-4, or
T. gondii infection. For a control, 463 bp of β-actin was amplified with a primer set of 5'-ATG GAT GAT GAT ATC GCC GCG-3' (forward) and 5'-AGT CCA TCA CGA TGC CAG TGG-3' (reverse) [
14]. The density of amplified DNA was measured by Image capture and analysis software in Eagle Sight TM 3.0 (Stratagene, La Jolla, California, USA).
The promoter region of Eotaxin-3 was amplified with a primer set of 5'-TGA CTC GAG TCT GTT AGA TCT CTC AAA TGC C-3' (forward) and 5'-TCA AGC TTC ATC ATG ATG CTG CAA ATC AG-3' (reverse) from genomic DNA of HeLa cells isolated by TRIzol reagent (Invitrogen) [
13]. The amplified 970 bp Eotaxin-3 promoter region was cloned in pGL3-basic (Promega, Madison, Wisconsin, USA). After co-transfection with pGL3-Eotaxin-3 and β-gal vector, cells were stimulated with 50 ng/ml IL-4, 100 ng/ml IFN-γ, or 1 × 10
8 T. gondii tachyzoites. Cells were lysed after 24 hr incubation, and then subjected to a luciferase assay system (Promega) and β-galactosidase enzyme assay system (Promega). Both assays were measured by a VICTOR3™ 1420 multilabel counter (Perkin Elmer, Boston, Massachusetts, USA).
Data were presented as the mean ± SD and significant differences were assessed using the paired Student's t test. P values less than 0.05 were considered statistically significant.
RESULTS
Nuclear translocation of STAT6 by T. gondii infection
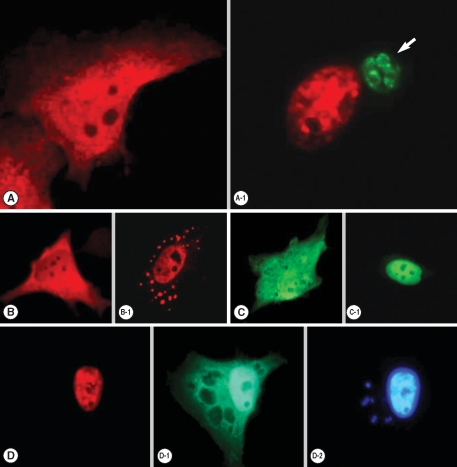
Transfected pDs2Red-STAT6 and pEGFP-STAT1α plasmids were expressed well to disperse red and green fluorescence each other in the cytoplasm of HeLa cells (
Fig. 1), which were translocated to the nucleus by the entry of
T. gondii (
Fig. 1A-1), after IL-4 treatment (
Fig. 1B-1) and IFN-γ treatment (
Fig. 1C-1). When the HeLa cells were co-transfected with both plasmids and then challenged with
T. gondii, all the STAT6 expressed as a cytosolic form of red fluorescence was observed in the infected HeLa cell nucleus (
Fig. 1D). Green fluorescence of STAT1α was observed in the nucleus partially and in the cytoplasm of the host cell as scattered fluorescence with hollows (
Fig. 1D-1). This hollows in the cytoplasmic fluorescence indicated the residence of
T. gondii within parasitophorous vacuolar membrane (PVM) which was observed as small blue spots of
T. gondii nuclei overlapping the hollows near a large host cell nucleus by staining with Hoechst 33258 (
Fig. 1D-2).
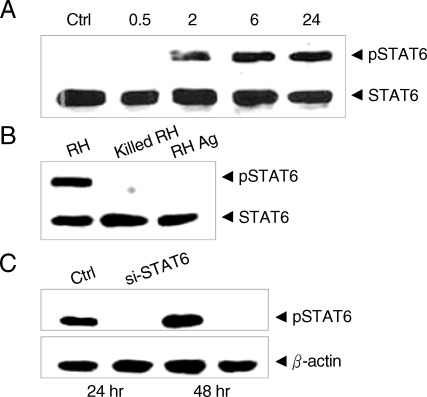
Nuclear translocation of STAT6 was confirmed to be the result of phosphorylation of the STAT6 by
T. gondii infection in the Western blot analysis. Phosphorylation of STAT6 appeared as early as 2 hr after infection of
T. gondii and reached a plateau in 6 hr sufficient time for the PVM maturation (
Fig. 2A). There were no detectable changes in the cytoplasmic STAT6 pool regardless of transition of STAT6 to phospho-STAT6. The phosphorylation of STAT6 occurred only when the parasites penetrated into host cells and did not occur with the killed tachyzoites or with soluble
T. gondii extracts (
Fig. 2B). It was inhibited completely by the treatment of siSTAT6 from 24 hr and maintained to 48 hr (
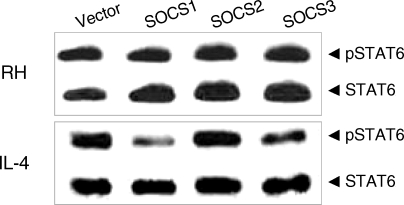
Fig. 2C). The activation of STAT6 by
T. gondii infection was not inhibited by any excess SOCS's whereas activation by IL-4 was inhibited by SOCS-1 and SOCS-3 (
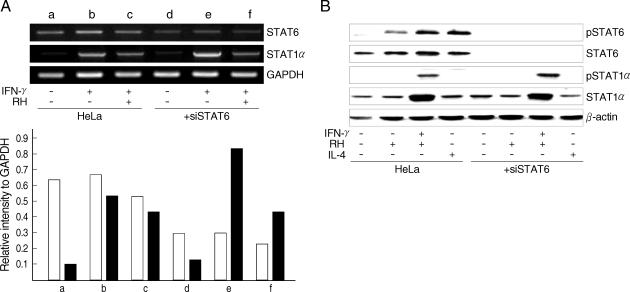
Fig. 3). The level of STAT6 transcription was reduced but that of STAT1α was increased highly with IFN-γ treatment under the siSTAT6 incubation for 24 hr whereas the STAT1α transcription was reduced more rapidly by
T. gondii infection (
Fig. 4A). These were reflected in the level of phosphorylation of STAT6 and STAT1α in western blot (
Fig. 4B).
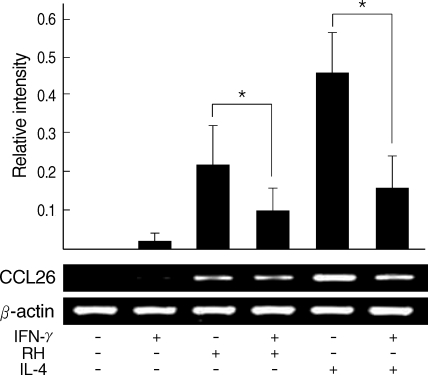
Eotain-3 gene expression was chosen as a marker for the action of activated STAT6.
T. gondii infection induced Eotaxin-3 gene expression which was reduced by treatment with IFN-γ. IL-4 also induced Eotaxin-3 expression, which was inhibited by IFN-γ (
Fig. 5). IL-4-like action of
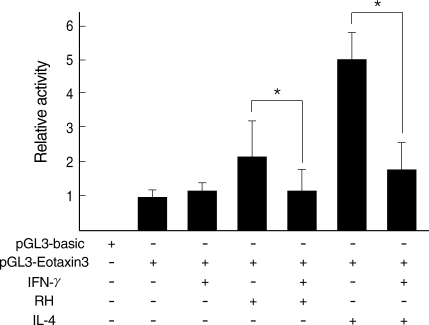
T. gondii infection against IFN-γ was further confirmed by a promoter assay of Eotaxin-3 gene (
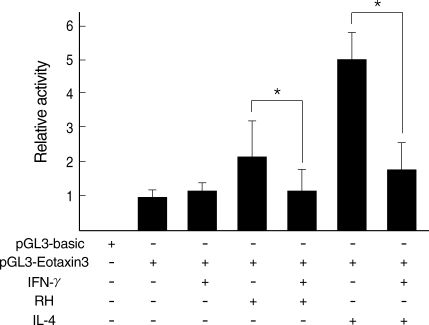
Fig. 6). Luciferase activity was increased with
T. gondii infection and decreased by IFN-γ treatment as the similar pattern as induced by IL-4 and then inhibited by IFN-γ. Reduction of Eotaxin-3 gene expression by IFN-γ was statistically significant in both assays between
T. gondii infection group and IL-4 treatment group.
DISCUSSION
We showed in this study that
T. gondii infection exploits STAT6 activation to modulate microbicidal action of IFN-γ in infected cells. Activation of STAT6 was accomplished within 2 hr after infection, and was not observed with killed tachyzoites or soluble
T. gondii extracts. Until now STAT6 has been known as unique signaling mediator of IL-4 and IL-13, which share many biological functions with IL-4 [
15]. Therefore, we have focused on the responses of the infected host cells that received IL-4-like signaling when
T. gondii penetrated into the cells.
There is little information on the participation of IL-4 in the immune modulation by
T. gondii infection and this is likely because there are no symptoms related to IL-4 such as asthma or eosinophilia after infection with
T. gondii. During the course of
T. gondii infection, IL-4 exerts a protective effect in the early acute phase but is detrimental over the long term infection [
16]. In IL-4 knockout mice, mortality during the early acute phases of
T. gondii infection was reduced due to the down-regulatory effects of IL-4, or associated Th2-derived products of proinflammatory cytokines such as IFN-γ. However, in long term infections, IL-4 increased parasite multiplication outside of cysts, which resulted in the development of severe necrotic cerebral lesions. Strain-specific differences in susceptibility to
T. gondii infection have demonstrated that the survival rate was significantly reduced in disease-susceptible mice deficient in IL-4 while those with
T. gondii-resistant strain did not have survival reduction [
17]. These results suggest participation of IL-4 in the body's immune system response to
T. gondii infection.
At the cellular level, IFN-γ exerts its effects on the elimination of intracellular
T. gondii through microbicidal compounds such as production of nitric oxide (NO) [
18], activation of IFN-inducible GTPase (IGTP) [
19], and production of LRG-47 [
20]. On the while IL-4 regulates NO production through depletion of arginine, the substrate of inducible NO synthase activated by IFN-γ to kill the intracellular
T. gondii [
21]. IL-4, and IL-13 also, down-regulates indoleamine 2,3-dioxygenase (INDO) activity, which catalyzes tryptophan that is an essential amino acid for
T. gondii growth within the host cells, activated by IFNs especially by IFN-γ [
22,
23]. Moreover, IFN-γ directly restricts the IL-4 action via inhibition of STAT6 signal transduction and gene expression [
24] or by suppression of STAT6 phosphorylation by inhibiting its recruitment to the IL-4 receptor [
25]. Intracellular infection of
T. gondii leads to a blockade in NFκB and STAT1 activation pathways. While IκB is phosphorylated and degraded, and STAT1 undergoes phosphorylation-dependent activation, neither NFκB nor STAT1 translocate into the nucleus during early infection of
T. gondii [
26]. Therefore, production of IL-12 and TNF-α is suppressed, and expression of MHC molecules is down-regulated [
27].
T. gondii infection also renders cells resistant to apoptosis, as shown by reduced caspase proteolytic activation and mitochondrial cytochrome c release [
28]. IL-4 rescues the cells from apoptosis with the cooperation of insulin receptor substrate-2 (IRS-2) [
29] and protects against NO induced apoptosis [
30]. Anti-apoptotic activity during the
T. gondii infection may be derived from STAT6 activation, which mimics IL-4 responses without adding actual exogenous IL-4. Gene expressional interaction between STAT6 and STAT1α was inhibitory each other as revealed in
Fig. 4.
When the IL-4 receptor is stimulated by IL-4, cytosolic STAT6 is phosphorylated via receptor-associated Janus kinase (Jak) 1 or 3 to make dimers [
31]. A dimer of phosphotyrosine-STAT6 translocated to the nucleus binds to p100, a co-activator for STAT6 to transcribe IL-4-induced genes, to bridge with RNA polymerase II [
32] and to recruit histone acetyltransferase activity to STAT6 to mediate interaction between the CREB-binding protein and STAT6 [
33]. It is unclear, however, from the results of this study, which Jak participates in the phosphorylation of cytosolic STAT6 during the entry and growth of
T. gondii in host cells. As revealed by STAT6 activation as early as within 2 hr (
Fig. 2), Jak1, Jak3, or other Jak-like kinase might apposition to the receptor-like molecules on the primary parasitophorous vacuolar membrane (PVM) to activate STAT6. Translocated to the nucleus, activated STAT6 binds to the γ-activated site (GAS) family of enhancers specific for IL-4. Among them, gene expression of Eotaxin-3 (CCL26), a potent chemoattractant for eosinophils, is known as a marker for IL-4 action through STAT6 signaling [
13,
14]. Quantitative RT-PCR of Eotaxin-3 gene expression demonstrated that Eotaxin-3 was induced by
T. gondii infection, and that the expression was reduced by IFN-γ treatment (
Fig. 5). This was further confirmed by a luciferase assay of the promoter region of the Eotaxin-3 gene (
Fig. 6). The differences observed in Eotaxin-3 gene expression between
T. gondii infection group and IL-4 treatment group were caused by non-infected host cells in the
T. gondii infection group, but reduction of Eotaxin-3 expression by IFN-γ treatment showed statistical significance in both groups.
Cytokine signaling is stopped by SOCS which is rapidly upregulated after stimulation with a cytokine, in case with IL-4 stimulation, SOCS-1 and SOCS-3 function to decay the stimuli [
34].
T. gondii-induced phosphorylation of STAT6 was maintained regardless of SOCS's which were over-expressed by transfected SOCS plasmids while that associated with IL-4-induction was reduced by SOCS-1 and SOCS-3. This is similar to the case of STAT3 activation by
T. gondii infection [
9], but different from cases of STAT1 activation by
T. gondii infection and by
Chlamydia trachomatis infection where the activation of STAT1 is dependent on bacterial replication within the host cells [
35]. The sustained STAT activation during the infection period reflects the continuous activation of STAT by the residing pathogens to cope with the host's attack of microbicidal effects. Furthermore, any SOCS's can not affect the decay of STAT activation during this process.
Without stimulating the IL-4 secreting cells such as Th2 cells, mast cells, and NK cells, T. gondii infection itself triggers the autonomous STAT6 activation pathway to obstruct the innate immunity of IFN-γ produced by the infection. This serves as a minimal strategy for the growth and replication of T. gondii within the host cells. It will be helpful to understand the parasitism of T. gondii which molecule in the PVM phosphorylates STAT6 specifically and how to inhibit the phosphorylation of STAT6 or the nuclear translocation of STAT6.
ACKNOWLEDGEMENTS
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-311-E00262).
References
Fig. 1Fluorescent localization of transfected STAT6 and STAT1α in HeLa cells. (A) Expression of pDsRed2-STAT6 plasmid was dispersed as a cytosolic form, which localized (A-1) nucleus after phosphorylation in the T. gondii infected (arrow) host cell. (B) Expression of pDsRed2-STAT6 product in the cytoplasm then moved to the nucleus after IL-4 treatment (B-1), (C) expression of pEGFP-STAT1α plasmid as a cytosolic form and (C-1) nuclear translocation of pEGFP-STAT1α product after IFN-γ treatment. When pDsRed2-STAT6 and pEGFP-STAT1α plasmids were co-transfected, (D) STAT6 was observed in the nucleus after T. gondii infection, (D-1) STAT1α in the nucleus and partially cytoplasm (D-2) as confirmed by the nuclear staining with Hoechst 33258 dye as a large nucleus of the host cell and several fluorescent spots from the growing T. gondii nuclei (arrow) within the cytoplasm of the host cell.
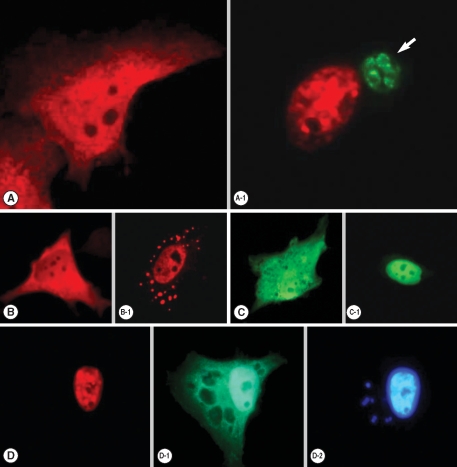
Fig. 2Western blot of T. gondii infected HeLa cell extracts with monoclonal anti-STAT6 and anti-phosphotyrosine STAT6 antibodies. (A) HeLa cells were harvested on the time indicated (hr) after infection with T. gondii (Ctrl, uninfected control) (B) HeLa cells were harvested after infection with T. gondii (RH), killed tachyzoites (killed RH), and soluble extracts (RH Ag), and (C) phosphorylation was inhibited by siSTAT6 for 24 or 48 hr before the infection with T. gondii, respectively.

Fig. 3Effects of the suppressors of cytokine signaling (SOCS) on the activation of STAT6 by T. gondii infection. SOCS-1, -2, and -3 were transfected for expression and then T. gondii infected (RH) or IL-4 treated (IL-4) for Western blot analysis.
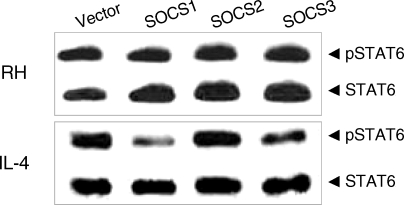
Fig. 4Inhibitory effects of STAT6 and STAT1α in (A) transcriptional level and (B) in protein expressional level by T. gondii infection through siSTAT6 treatment. White rectangles indicated STAT6 and blacks STAT1α.

Fig. 5
Quantitative RT-PCR of Eotaxin-3 gene expression after T. gondii infection. Relative intensity of Eotaxin-3 to β-actin was calculated.
*Statistical significance by the paired Student's t test (P < 0.05).

Fig. 6
Luciferase assay of Eotaxin-3 promoter region by T. gondii infection. Relative luciferase activity was estimated to that of the vector (pGL3-basic) with promoter region (pGL3-Eotaxin-3).
*Statistical significance by the paired Student's t test (P < 0.05).