Abstract
Intestinal giant-cystic disease (IGCD) of the Israel carp (Cyprinus carpio nudus) has been recognized as one of the most serious diseases afflicting inland farmed fish in the Republic of Korea, and Thelohanellus kitauei has been identified as the causative agent of the disease. Until now, studies concerning IGCD caused by T. kitauei in the Israel carp have been limited to morphological and histopathological examinations. However, these types of diagnostic examinations are relatively time-consuming, and the infection frequently cannot be detected in its early stages. In this study, we cloned the full-length 18S rRNA gene of T. kitauei isolated from diseased Israel carps, and carried out molecular identification by comparing the sequence with those of other myxosporeans. Moreover, conventional PCR and real-time quantitative PCR (qPCR) using oligonucleotide primers for the amplification of 18S rRNA gene fragment were established for further use as methods for rapid diagnosis of IGCD. Our results demonstrated that both the conventional PCR and real-time quantitative PCR systems applied herein are effective for rapid detection of T. kitauei spores in fish tissues and environmental water.
-
Key words: Thelohanellus kitauei, Cyprinus carpio nudus, intestinal giant-cystic disease, identification, 18S rRNA, quantitative PCR (qPCR)
INTRODUCTION
Intestinal giant-cystic disease (IGCD) induced by the myxozoan parasite
Thelohanellus sp. was first reported in common carps,
Cyprinus carpio, in Japan [
1]. The causative agent,
Thelohanellus kitauei, was subsequently identified as a new species due to its specific balloon-like spore sack and intestinal parasitic habitat by Egusa and Nakajima [
2]. They also described morphological characteristics of
T. kitauei spores and salient pathological changes in common carps [
2]. In the Republic of Korea (=South Korea),
T. kitauei infection was first reported in the farmed Israel carp (
Cyprinus carpio nudus), and IGCD caused by
T. kitauei has been recognized as one of the most serious diseases occurring in inland farmed fish since 1988 [
3]. Infected fish evidence profound intestinal swelling and become emaciated due to the blockage of the intestinal tract by giant cysts, and the fish often ultimately die from the resultant enteritis [
2,
3]. Moreover, in cases of IGCD outbreaks, mortality tends to be focused largely in adult (2- and 3-year-old) Israel carps, and can reach levels as high as 10%. Mortality rates such as this can prove to be catastrophic and could severely damage the aquaculture industry [
2-
5].
No detailed information is currently available regarding transmission mechanisms and the life cycle of
T. kitauei, although the life cycle of
T. kitauei may be similar to that of other myxosporean parasites, including an actinosporean stage in an alternate oligochaete host. Among
Thelohanellus spp., the life cycles of
T. hovorkai and
T. nikolskii, which consist of myxosporean and actinosporean stages, have been confirmed at the molecular level, and the transmission and production of actinospores using the alternate oligochaete host have been experimentally demonstrated [
6-
9]. The high prevalence of actinosporean infections in oligochaete hosts such as
Branchiura sowerbyi has been suggested to be one of the etiological factors responsible for disease outbreaks [
10].
Although IGCD outbreaks and mortality in the Israel carp have occurred continuously and periodically since the first report of the disease in 1988, no detailed systematic strategies for its control have yet been developed. Only the antibiotic fumagillin has currently been proved as an effective drug for the treatment of IGCD in South Korea by Rhee et al. [
11]. They pointed out that fumagillin should be administered before the disease develops, as the efficacy of the drug is limited only to the early stages of infection. Thus, continuous monitoring of the disease and parasite spores in fish and environmental water should be implemented in order to maximize the drug efficacy. Classical diagnostic methods such as microscopic and histopathological examinations are time-intensive, and have proven incapable of detecting the infection in its early stages, during which fish do not yet show any gross signs of disease, including cyst formation. Therefore, development of more rapid methods of disease diagnostics and a systematic survey system capable of detecting the early stages of infection is urgently needed to enhance the efficacy of treatment and prevent the diffusion of IGCD in the Israel carp.
Recently, real-time quantitative PCR (qPCR) has become a valuable tool for the estimation of the parasite load, infection course, and environmental monitoring of certain myxozoan parasites, including
Parvicapsula minibicornis and
Myxobolus cerebralis [
12-
16].
Therefore, in this study, conventional PCR and real-time qPCR were applied for the detection of T. kitauei infections and quantification of infection intensity, as part of a broader effort to develop new methods for rapid diagnosis of IGCD in the Israel carp. Because relatively little information is available regarding the genetic features of T. kitauei, the genetic identification of T. kitauei in the Israel carp via nucleotide sequencing of the 18S small subunit ribosomal RNA (18S rRNA) gene was conducted prior to PCR detection. In an effort to evaluate the infection and host specificity, PCR detection of T. kitauei was conducted using freshwater fish cultivated in the same environment as the Israel carp, a species which is particularly vulnerable to T. kitauei infection.
MATERIALS AND METHODS
Isolation of parasite spores
Giant egg-shaped cysts were isolated from the intestines of diseased Israel carps grown in a pond aquarium located in Gochang, Jeonbuk Province, the west region of South Korea, from July to August, 2008. After then, we have continually isolated the cysts from diseased Israel carps until July, 2010. After cutting the cysts into small pieces with a pair of scissors, the cysts removed from the infected fish intestine were resuspended and diluted in sterile 0.9% physiological saline (PS) for morphological observations. The spore suspensions in 0.9% PS were kept in 4℃. The size and morphological characteristics of the spores (at least 50 spores per fish) were evaluated under an optical microscope (Carl Zeiss, Oberkochen, Germany).
Molecular identification of parasite species
Genomic DNA of mature spores was prepared via the following procedure. In brief, isolated mature spores were suspended in 500 µl of lysis buffer (10 mM Tris/pH 8.0, 100 mM NaCl, 7.5 mM EDTA, 1% SDS), and incubated for 1 hr at 68℃. DNA was subsequently purified using the High Pure PCR template preparation kit (Roche, Mannheim, Germany) in accordance with the manufacturer's protocols. The 18S small subunit ribosomal RNA (18S rRNA) was amplified by PCR using a set of universal eukaryotic primers (UEP-F, 5'-ACCTGGTTGATCCTGCCAG-3' and UEP-R, 5'-CTTCCGCAGGTTCACCTACGG-3', [
17]). PCR was conducted using a mixture containing 100 ng of genomic DNA, 10 µM of each primer, 2.5 mM dNTP mix, 2.5 units of Taq polymerase (Promega, Madison, Wisconsin, USA) and the manufacturer's reaction buffer in a final volume of 50 µl under the following conditions: 94℃ for 7 min (initial denaturation), followed by 30 cycles of 94℃ for 30 sec (denaturation), 55℃ for 30 sec (annealing), and 72℃ for 1 min (extension), and a final extension of 72℃ for 10 min. The PCR products were then excised from 1% agarose gels, and purified PCR products were cloned into pCR 2.1-TOPO vectors using the TOPO TA cloning kit (Invitrogen; Carlsbad, California, USA). The nucleotide sequences of clones (named pKitauei) of the PCR products were determined via sequencing. DNA sequencing was conducted with M13 Forward/Reverse primers using an ABI3730 automatic sequencer (96-capillary, Applied Biosystems, Foster City, California, USA) and Applied Biosystems BigDye® Terminator Cycle Sequencing Kits v3.1, in accordance with the manufacturer's recommendations. Finally, the obtained nucleotide sequence was compared with the 18S rRNA sequences of other myxosporeans in Genbank database via alignment using the CLUSTAL W software.
The full-length 18S rRNA gene-harboring plasmid (pKitauei), the suspension of spores, genomic DNA from isolated spores, and genomic DNA from the spore-spiked tissues were used as templates for both conventional PCR and real-time PCR assays. The pKitauei plasmid was used to make the standard curve for calculation of the copy number. For the reactions, the plasmid DNA was serially diluted 10-fold from 103 pg to 10-5 pg per µl. Spore suspensions containing 0, 100, 1,000, 10,000, 100,000, and 1,000,000 spores per ml nuclease-free water were prepared by 10 min of boiling at 98℃, and 5 µl of each prepared T. kitauei spore suspension was used directly in PCR. The genomic DNA of the isolated spores was prepared via purification of genomic DNA from spore suspensions containing 0, 1, 5, 10, 50, 100, 500, 1,000, 5,000, 10,000, 50,000, 100,000, and 500,000 spores using the High Pure PCR template preparation kit. The final elution volume of purified genomic DNA was adjusted to 100 µl, and 1 µl of DNA elute was used in the following PCR. The genomic DNA of the spore-spiked tissues was prepared by purifications of genomic DNA from mixtures of 0, 1, 10, 100, 1,000, and 10,000 spore-containing 10 µl of PS and 30 mg of small pieces of spore-free intestinal tissues using the High Pure PCR template preparation kit. The concentration of the purified DNA was determined using a NanoDrop spectrophotometer and 100 ng of DNA was used in the following PCR.
Detection and quantification of parasite spores via conventional PCR and real-time qPCR
For early detection and quantification of the infection, the detection limit of parasites was determined using conventional PCR and real-time qPCR assays using templates prepared above and following primers. The forward primer TkF-1: 5'-GCCCAGTAATCTACTATTCGACG-3' and the reverse primer TkR-1: 5'-GCTATTGATCTGTTAATCCTATC-3' were used to amplify the 716 bp fragment of
T. kitauei 18S rRNA gene. Conventional PCR was conducted using AccuPower™ PCR PreMix (Bioneer, Seoul, Korea) for the common laboratory experiments, and real-time qPCR was carried out using the FastStart Universal SYBR Green Master (Roche) for quantification of the infection intensity. The thermal cycler and real-time qPCR system used in this study were an iCycler and an IQ™5 Multicolor Real-Time PCR Detection System (BioRad, Hercules, California, USA), respectively. Both conventional and real-time PCR were performed under the following conditions: 94℃ for 5 min, followed by various cycles of 94℃ for 30 sec, 53℃ for 30 sec, and 72℃ for 40 sec. The conventional PCR products were analyzed after 30 (spore suspensions, genomic DNA of spores and genomic DNA of spore-spiked tissues) or 35 cycles (pKitauei) by 1% agarose gel electrophoresis and ethidium bromide staining. The results of real-time PCR were expressed as Cq (quantification cycle) values proposed in the report by Bustin et al. [
18] after 40 cycles and the standard curves were generated from the Cq values of 10-fold serially diluted pKitauei plasmid and DNA purified from various numbers of spores.
To investigate the disease prevalence and host specificity, PCR detection of T. kitauei was carried out using some freshwater fish. The Israel carp (Cyprinus carpio nudus), colored carp (Cyprinus carpio haematopterus), crucian carp (Carassius carassius), Far Eastern catfish (Silurus asotus), and Korean bullhead fish (Pseudobagrus emarginatus and Pseudobagrus fulvidraco) were acquired from an aquarium in the Inland Aquaculture Research Center of the National Fisheries Research and Development Institute, South Korea. IGCD outbreak in the Israel carp often occurred in this place. Ten fish from each fish species were employed in this experiment. Genomic DNA was prepared from 50 mg of the foregut, midgut, and hindgut portions of the intestine using the High Pure PCR template preparation kit (Roche). PCR detection was carried out using 100 ng of purified genomic DNA, TkF-1 primer, TkR-1 primer, and AccuPower™ PCR PreMix (Bioneer). PCR products were analyzed via 1% agarose gel electrophoresis and ethidium bromide staining. The 716 bp PCR products from PCR samples were cloned and sequenced to confirm the exact nucleotide sequences.
RESULTS
Characterization of spores
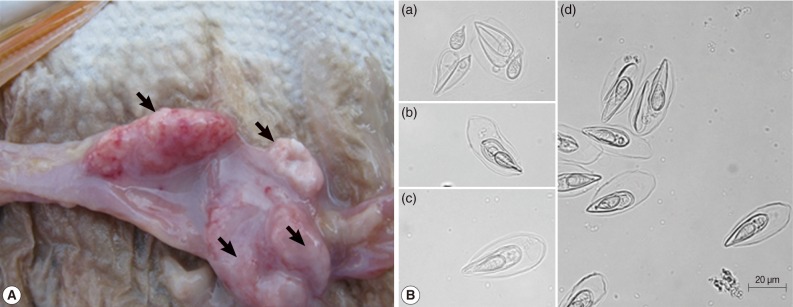
The egg-shaped giant cysts (12-45 mm size) removed from the intestines of infected Israel carps were filled with numerous mature spores (
Fig. 1A). The spores released from the cyst evidenced the typical specific features of
T. kitauei, including an egg-shaped balloon-like sack. The size of spores, spore sacks, polar capsules, and polar filament were identified in accordance with the descriptions provided by Egusa and Nakajima [
2] and Chun et al. [
3]. We also observed spores in front (
Fig. 1Bc), spores in side view (
Fig. 1Bb), opened spore sacks, and divided spores within the sack (
Fig. 1Ba, d) as previously described [
2].
The 2,048 bp of the full-length 18S rRNA gene was PCR amplified using the genomic DNA of the spores and the universal primer sets of UEP-F and UEP-R. After the analysis of the DNA sequences of PCR products from 10 samples, the isolated spores were identified as originating from a single species. The sequence obtained from this study has been deposited in the GenBank database (
http://www.ncbi.nlm.nih.gov/nucleotide) under the accession number HM624024. The nucleotide sequence of the
T. kitauei 18S rRNA gene obtained in this study was compared with those of other myxosporean parasites deposited previously in the GenBank using the CLUSTAL W version 1.8, as implemented in the BioEdit sequence alignment program. The
T. kitauei 18S rRNA gene in this study evidenced a sequence identity of 99.7% with the partial sequence of
T. kitauei 18S rRNA previously deposited by other authors (GenBank accession no. GQ396677.1). Additionally, the sequence showed high levels of identity with other myxosporeans, including
Thelohanellus spp.:
Thelohanellus hovorkai (97.0%; GenBank no. DQ231155.1),
T. hovorkai (95.0%; GenBank no. DQ445295.1),
T. hovorkai (95.0%; GenBank no. AJ133419.1),
Thelohanellus nikolskii (89.1%; GenBank no. GU165832.1),
Thelohanellus wuhanensis (91.9%; GenBank no. AY165181.1),
Thelohanellus sinensis (88.3%; GenBank no. DQ452013.1),
Thelohanellus zahrahae (78.8%; GenBank no. EU643622.1),
Hexaactinomyxon sp. (87.0%; GenBank no. DQ473517.1), and
Myxobolus turpisrotundus (85.1%; GenBank no. EF690299.1).
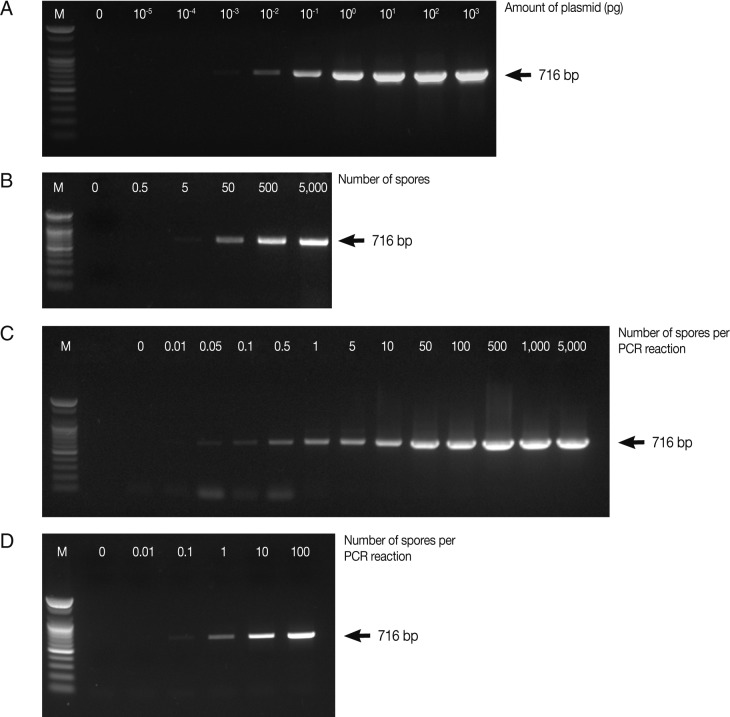
In this study, for early detection of the infection status, the detection limits of parasites were determined by PCR and real-time qPCR. PCR was carried out using various concentrations of the full-length 18S rRNA gene-harboring plasmid (pKitauei), the suspension of spores, genomic DNA from isolated spores, and genomic DNA from the spore-spiked tissues as templates. We initially carried out conventional PCR and real-time qPCR using different concentrations of the pKitauei plasmid for further use to estimate the DNA amount of
T. kitauei. Conventional PCR was capable of detecting 10
-3 pg of the pKitauei plasmid within only 30 cycles (
Fig. 2A) and 10
-4 pg of plasmid DNA could be detected over 35 cycles of real-time qPCR (
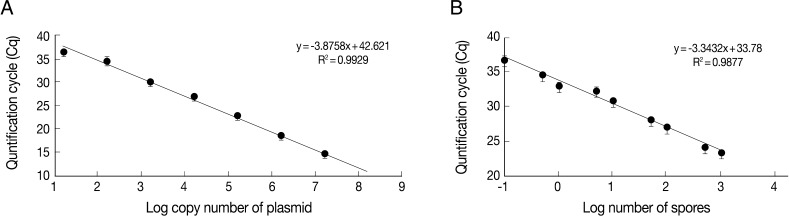
Table 1). Linear standard curves could be generated by real-time qPCR from the Cq values of the diluted plasmid pKitauei and DNA purified from various numbers of spores (
Fig. 3) and primer specificity was confirmed via melt-curve analysis in all samples (data not shown).
When the spore suspensions were used as templates, as few as 5 spores could be detected by conventional PCR (
Fig. 2B), whereas the amplification of parasite DNA from spore suspensions failed, and no detectable Cq values were obtained by real-time PCR after 40 cycles. In this study, when genomic DNA from various numbers of isolated spores were used, the PCR reactions contained genomic DNA of 0, 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 50, 100, 500, 1,000, and 5,000 spores after dilution. As a consequence, very small amounts of parasite genomic DNA, i.e., equivalent to 0.05 spore and 0.1 spore, could be detected by conventional PCR and real-time PCR, respectively (
Table 1;
Fig. 2C). Finally, conventional and real-time PCR assays using genomic DNA prepared from the spore-spiked tissues as templates were carried out to determine whether the detection systems employed in this study were applicable to estimate the intensity of infection in fish tissues. Positive reactions were noted in conventional and real-time PCR when the fish genomic DNA containing DNA of more than 0.1 or 1 spores was used as a template for conventional PCR or real-time PCR, respectively (
Table 1;
Fig. 2D).
In this study, the conventional PCR detection of T. kitauei using the primer set designed in this study was performed using several freshwater fish such as the colored carp (C. carpio haematopterus), crucian carp (C. carassius), Far Eastern catfish (S. asotus) and Korean bullhead fish (P. emarginatus and P. fulvidraco), in order to evaluate the infection prevalence and host specificity. As results of 1% agarose gel electrophoresis analyses, 100% positive PCR reactions were noted in the intestines of examined fish species. The nucleotide sequences of the cloned positive PCR products matched the sequence of T. kitauei, and the sequence identity was 99.4-100%.
DISCUSSION
In this study, we cloned the full-length 18S small subunit ribosomal RNA (18S rRNA) gene of T. kitauei isolated from the diseased Israel carp C. carpio nudus, and molecular identification was conducted via comparisons of this sequence with those of other myxosporeans, including the previously submitted T. kitauei sequence. Moreover, conventional and real-time quantitative PCR methods were established and the detection limits of both PCR systems were evaluated to quantify the infection intensity. Our findings showed that both conventional and real-time PCR techniques were capable of detecting very small amounts of spore DNA, i.e., less than 1 spore, in various types of samples. Although conventional PCR is more efficient than real-time PCR, the detection limits of both PCR systems were similar. This indicates the sub-optimal efficiency of the real-time PCR assay or different sensitivities of taq polymerases between the 2 PCR systems. In future studies, we wish to further establish optimal conditions for improving real-time PCR efficiency and specificity. According to the combined results of conventional and real-time PCR, approximately 1.5×102 copies of the 18S rRNA gene were present in a single mature spore.
Pathogen monitoring is an integral part of developing management strategies to reduce the impact of disease on free-ranging and wild fish populations as well as farmed fish populations [
15], and real-time PCR has been used as a useful tool for the detection and quantification of a variety of myxozoan parasites [
12,
13]. Moreover, the number of water-borne parasites in water samples may be considered an indirect indicator of infection risk or intensity, and real-time PCR analysis of water samples may prove useful as a novel method to replace the traditional use of susceptible host fish for both the detection and quantification of specific parasites [
15,
16,
19-
21]. In this study, as a preliminary study for pathogen monitoring of environmental water, DNA prepared from spore-suspensions was used in PCR assays. However, further studies detecting spore DNA using real water samples are required to demonstrate this method as a practical protocol. We have used only mature spores as materials for our detection experiments, because the actinospores of
T. kitauei have not yet been identified. Notwithstanding, the methods established herein should prove to be useful for further studies directly addressing the actinosporean stage of the parasite, since DNA is conserved throughout the life cycle of the parasite. The conventional PCR system used in our study detected spore DNA from spore suspensions without the need for genomic DNA preparation procedures, although real-time PCR systems were unable to detect spore DNA in the spore suspensions. This might be attributable to different sensitivities between the taq polymerase activities of the 2 PCR systems, although we suggest that a genomic DNA preparation step should be included before PCR in order to detect
T. kitauei spores with greater sensitivity.
In this study and previous studies,
T. kitauei has been clearly discriminated from other
Thelohanellus sp., including
T. hovorkai, according to the morphology of spores and the specific infection site [
2,
3]. However, the level of exhibited sequence identity of 18S rRNA sequence with
T. hovorkai was quite high (97%). The PCR assay established in this study may be sufficient to detect
T. kitauei spores in case of the Israel carp, although similar myxosporean spores, including
T. hovorkai may co-exist in wild streams. Therefore, further studies developing more specific primers or probes should be conducted for rapid detection by conventional or real-time qPCR without time-consuming cloning and sequencing procedures. As mentioned in some previous studies [
22,
23], the combined analyses of other appropriate genes, including mitochondrial sequences as well as 18S rRNA gene sequences might be useful to determine the precise phylogenetic position of
T. kitauei using comparison of these gene sequences and to enable more discerning diagnoses of diseases while avoiding the risk of false conclusions associated with traditional morphological methods [
12,
24-
27].
In an effort to evaluate the infection prevalence and possible host specificity of IGCD, the detection of
T. kitauei via the conventional PCR method established herein was carried out using some freshwater fish cultivated using the same environmental water with the Israel carp, a species which is known to be particularly vulnerable to
T. kitauei infection. As a consequence, almost all of the tested fish harbored
T. kitauei spores, although no disease outbreaks or mortality have been reported thus far with the exception of the Israel carp. There are several possibilities; the parasite could indeed have a broad host range, or because the tissue tested is the intestine and the amount of detected spore DNA was relatively low, it could be that these fish have simply ingested parasites. Although similar results and suggestions on the fish susceptibility to other myxozoan parasites such as
T. nikolskii and
Myxobolus cerebralis have been reported by several researches [
14,
28-
31], very little information is currently available regarding exact mechanisms underlying the myxozoan host specificity in fish. Therefore, further studies are required on the correlation between the relative infection intensity and disease development in other fish species as well as the Israel carp.
This is the first study, to the best of our knowledge, to detect and quantify T. kitauei spores by PCR methods in the Israel carp. However, even after this study, a great many questions about T. kitauei might persist. In future studies, using these detection systems and also more advanced systems, we will investigate in greater detail the various stages of the life cycle by surveying different morphotypes of T. kitauei in fish, alternate hosts, and environmental water at the molecular level, in an effort to evaluate and clarify the exact mechanisms of infection and life cycle characteristics of T. kitauei.
ACKNOWLEDGMENT
This study was supported by a research fund of the National Fisheries Research and Development Institute (NFRDI), the Republic of Korea.
References
- 1. Kitaue K. Intestinal giant-cystic disease affecting the carp, caused by Thellohanellus sp. Fish Pathol 1980;14:145-146.
- 2. Egusa S, Nakajima K. A new Myxozoa Thelohanellus kitauei, the cause of intestinal giant cystic disease of carp. Fish Pathol 1981;15:213-218.
- 3. Chun SK, Choi DL, Park IS. Studies of Thelohanellus infection in carp, Cyprinus carpio. I. Experimental induction of Thelohanellosis. J Fish Pathol 1988;1:111-116.
- 4. Rhee JK, Kim JO, Kim PG, Park BK. Prophylactic and therapeutic studies on intestinal giant-cystic disease of the Israel carp caused by Thelohanellus kitauei. I. Course of formation and vanishment of the cyst. Korean J Parasitol 1990;28:183-194.
- 5. Rhee JK, Kim JO, Park BK. Prophylactic and therapeutic studies on intestinal giant-cystic disease of the Israel carp caused by Thelohanellus kitauei. II. Effects of physical and chemical factors on T. kitauei spores in vitro. Korean J Parasitol 1990;28:241-252.
- 6. Yokoyama H. Transmission of Thelohanellus hovorkai Achmerov, 1960 (Myxosporea: Myxozoa) to common carp Cyprinus carpio through the alternate oligochaete host. Syst Parasitol 1997;36:79-84.
- 7. Szekély C, El-Mansy A, Molnar K, Baska F. Development of Thelohanellus hovorkai and Thelohanellus nikolskii (Myxosporea: Myxozoa) in oligochaete alternate hosts. Fish Pathol 1998;33:107-114.
- 8. Anderson CL, Canning EU, Schafer SM, Yokoyama H, Okamura B. Molecular confirmation of the life cycle of Thelohanellus hovorkai Achmerov, 1960 (Myxozoa: Myxosporea). Bull Eur Assoc Fish Pathol 2000;20:111-115.
- 9. Liyanage YS, Yokoyama H, Wakabayashi H. Dynamics of experimental production of Thelohanellus hovorkai (Myxozoa: Myxosporea) in fish and oligochaete alternate hosts. J Fish Dis 2003;26:575-582.
- 10. Yokoyama H, Liyanage YS, Sugai A, Wakabayashi H. Hemorrhagic thelohanellosis of color carp caused by Thelohanellus hovorkai (Myxozoa: Myxosporea). Fish Pathol 1998;33:85-89.
- 11. Rhee JK, Kim HC, Park BK. Efficacy of fumagillin against Thelohanellus kitauei infection of Israel carp, Cyprinus carpio nudus. Korean J Parasitol 1993;31:57-65.
- 12. Cavender WP, Wood JS, Powell MS, Overturf K, Cain KD. Real-time quantitative polymerase chain reaction (QPCR) to identify Myxobolus cerebralis in rainbow trout Oncorhynchus mykiss. Dis Aquat Organ 2004;60:205-213.
- 13. Kelley GO, Adkison MA, Zagmutt-Vergara FJ, Leutenegger CM, Bethel JW, Myklebust KA, McDowell TS, Hedrick RP. Evaluation of quantitative real-time PCR for rapid assessments of the exposure of sentinel fish to Myxobolus cerebralis. Parasitol Res 2006;99:328-335.
- 14. Kallert DM, Eszterbauer E, Grabner D, El-Matbouli M. In vivo exposure of susceptible and non-susceptible fish species to Myxobolus cerebralis actinospores reveals non-specific invasion behaviour. Dis Aquat Organ 2009;84:123-130.
- 15. Hallett SL, Bartholomew JL. Development and application of a duplex QPCR for river water samples to monitor the myxozoan parasite Parvicapsula minibicornis. Dis Aquat Organ 2009;86:39-50.
- 16. Griffin MJ, Pote LM, Camus AC, Mauel MJ, Greenway TE, Wise DJ. Application of a real-time PCR assay for the detection of Henneguya ictaluri in commercial channel catfish ponds. Dis Aquat Organ 2009;86:223-233.
- 17. Barta JR, Martin DS, Liberator PA, Dashkevicz M, Anderson JW, Feighner SD, Elbrecht A, Perkins-Barrow A, Jenkins MC, Danforth HD, Ruff MD, Profous-Juchelka H. Phylogenetic relationships among eight Eimeria species infecting domestic fowl inferred using complete small subunit ribosomal DNA sequences. J Parasitol 1997;83:262-271.
- 18. Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 2009;55:611-622.
- 19. Audemard C, Reece KS, Burreson EM. Real-time PCR for detection and quantification of the protistan parasite Perkinsus marinus in environmental waters. Appl Environ Microbiol 2004;70:6611-6618.
- 20. Hallett SL, Bartholomew JL. Application of a real-time PCR assay to detect and quantify the myxozoan parasite Ceratomyxa shasta in river water samples. Dis Aquat Organ 2006;71:109-118.
- 21. Bartholomew JL, Atkinson SD, Hallett SL, Zielinski CM, Foott JS. Distribution and abundance of the salmonid parasite Parvicapsula minibicornis (Myxozoa) in the Klamath River basin (Oregon-California, U.S.A.). Dis Aquat Organ 2007;78:137-146.
- 22. Fiala I. The phylogeny of Myxosporea (Myxozoa) based on small subunit ribosomal RNA gene analysis. Int J Parasitol 2006;36:1521-1524.
- 23. Eszterbauer E, Marton S, Rácz OZ, Letenyei M, Molnár K. Morphological and genetic differences among actinosporean stages of fish-parasitic myxosporeans (Myxozoa): difficulties of species identification. Syst Parasitol 2006;65:97-114.
- 24. Olson PD, Cribb TH, Tkach VV, Bray RA, Littlewood DT. Phylogeny and classification of the Digenea (Platyhelminthes: Trematoda). Int J Parasitol 2003;33:733-755.
- 25. Hypsa V, Skeríkova A, Scholz T. Phylogeny, evolution and host-parasite relationships of the order Proteocephalidea (Eucestoda) as revealed by combined analysis and secondary structure characters. Parasitology 2005;130(Pt 3):359-371.
- 26. Kim KH, Eom KS, Park JK. The complete mitochondrial genome of Anisakis simplex (Ascaridida: Nematoda) and phylogenetic implications. Int J Parasitol 2006;36:319-328.
- 27. Epp JK, Wood JS, Milton JB. New PCR test for Myxobolus cerebralis based on a heat-shock protein gene. Proceedings 8th Annual Whirling Disease Symposium. 2002. Denver, CO: 61-62.
- 28. Molnár K. Differences between the European carp (Cyprinus carpio carpio) and the coloured carp (Cyprinus carpio haematopterus) in susceptibility to Thelohanellus nikolskii (Myxosporea) infection. Acta Vet Hung 2002;50:51-57.
- 29. Baerwald MR, Welsh AB, Hedrick RP, May B. Discovery of genes implicated in whirling disease infection and resistance in rainbow trout using genome-wide expression profiling. BMC Genomics 2008;9:37.
- 30. el-Matbouli M, Hoffmann RW, Schoel H, McDowell TS, Hedrick RP. Whirling disease: host specificity and interaction between the actinosporean stage of Myxobolus cerebralis and rainbow trout Oncorhynchus mykiss. Dis Aquat Organ 1999;35:1-12.
- 31. Yokoyama H, Kim JH, Urawa S. Differences in host selection of actinospores of two myxosporeans, Myxobolus arcticus and Thelohanellus hovorkai. J Parasitol 2006;92:725-729.
Fig. 1Characterization of spores. (A) Various sizes of giant cysts (arrows) containing mature spores found in the intestine of an Israel carp infected with Thelohanellus kitauei. (B) Spores of T. kitauei isolated from the intestine of a diseased fish. Scale bar: 20 µm.
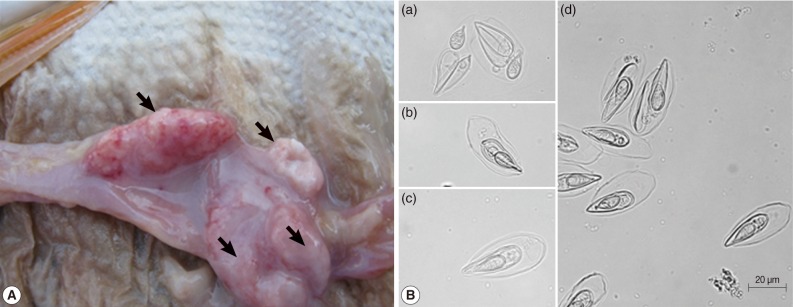
Fig. 2Determination of the detection limit of conventional PCR using AccuPower™ PCR PreMix system. Four µl of PCR products were loaded on 1% agarose gels and ethidium bromide-stained specific PCR bands were visualized using a UV-transilluminator. PCR was carried out using various concentrations of the full-length 18S rRNA gene-harboring plasmid pKitauei (A), the suspension containing different number of spores (B), genomic DNA purified from isolated spores (C) and genomic DNA from the various numbers of spore-spiked tissues (D) as templates. M: 100 bp ladder from
Bioneer.co.kr
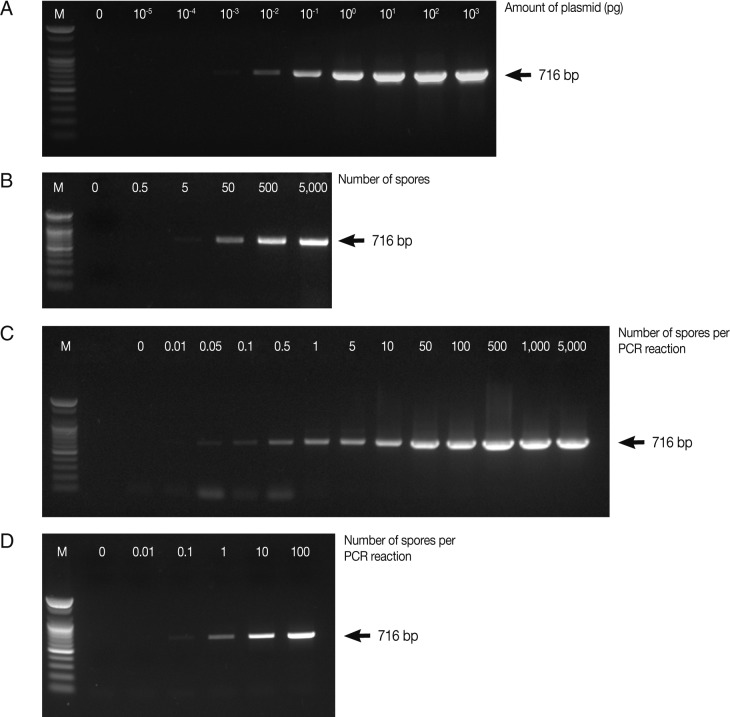
Fig. 3Linear standard curves derived from 10-fold serially diluted pKitauei plasmid (A) and DNA purified using known numbers of T. kitauei spores (B). x-axis and y-axis represent a log10 of number of spore used in DNA preparation and mean (± SD) real-time qPCR Cp value, respectively. PCR efficiency calculated using slope of the standard curve was 99.2%.
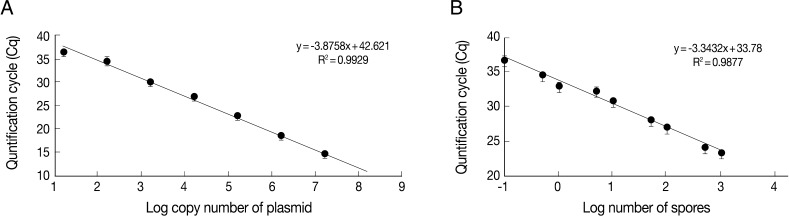
Table 1.Quantitative real-time PCR assay for detection of Thelohanellus kitauei. Mean Cq and SD values were obtained from triplicate runs
Table 1.
|
Samples |
Amount per reaction |
Mean Cq (SD) |
Samples |
Amount per reaction (no. of spores) |
Mean Cq (SD) |
Samples |
Amount per reaction (no. of spores) |
Mean Cq (SD) |
|
pKitauei |
103 pg |
9.8 (0.158) |
DNA from spore- spiked tissues |
100 |
32.16 (0.643) |
Genomic DNA from isolated spores |
5,000 |
21.75 (0.200) |
|
102 pg |
14.89 (0.290) |
|
10 |
35.61 (0.946) |
|
1,000 |
23.54 (0.147) |
|
101 pg |
18.79 (0.016) |
|
1 |
38.04 (0.305) |
|
500 |
24.27 (0.116) |
|
100 pg |
23.14 (0.040) |
|
0.1 |
N/A |
|
100 |
27.18 (0.185) |
|
10-1 pg |
27.27 (0.136) |
|
0.01 |
N/A |
|
50 |
28.22 (0.209) |
|
10-2 pg |
30.29 (0.136) |
|
0 |
N/A |
|
10 |
30.99 (0.284) |
|
10-3 pg |
34.75 (0.944) |
|
NTC |
N/A |
|
5 |
32.43 (0.516) |
|
10-4 pg |
36.62 (0.584) |
|
|
|
|
1 |
33.13 (0.473) |
|
10-5 pg |
N/A |
|
|
|
|
0.5 |
34.68 (0.189) |
|
0 |
N/A |
|
|
|
|
0.1 |
36.83 (0.589) |
|
NTC |
N/A |
|
|
|
|
0.05 |
N/A |
|
|
|
|
|
|
|
0.01 |
N/A |
|
|
|
|
|
|
|
0 |
N/A |
|
|
|
|
|
|
|
NTC |
N/A |