Abstract
Amoebic keratitis (AK) caused by Acanthamoeba is one of the most serious corneal infections. AK is frequently misdiagnosed initially as viral, bacterial, or fungal keratitis, thus ensuring treatment delays. Accordingly, the early detection of Acanthamoeba would contribute significantly to disease management and selection of an appropriate anti-amoebic therapy. Recently, the loop-mediated isothermal amplification (LAMP) method has been applied to the clinical diagnosis of a range of infectious diseases. Here, we describe a rapid and efficient LAMP-based method targeting Acanthamoeba 18S rDNA gene for the detection of Acanthamoeba using clinical ocular specimens in the diagnosis of AK. Acanthamoeba LAMP assays detected 11 different strains including all AK-associated species. The copy number detection limit for a positive signal was 10 DNA copies of 18S rDNA per reaction. No cross-reactivity with the DNA of fungi or other protozoa was observed. The sensitivity of LAMP assay was higher than those of Nelson primer PCR and JDP primer PCR. In the present study, LAMP assay based on directly heat-treated samples was found to be as efficient at detecting Acanthamoeba as DNA extracted using a commercial kit, whereas PCR was only effective when commercial kit-extracted DNA was used. This study showed that the devised Acanthamoeba LAMP assay could be used to diagnose AK in a simple, sensitive, and specific manner.
-
Key words: Acanthamoeba, loop-mediated isothermal amplification (LAMP), 18S rDNA, rapid detection
INTRODUCTION
Acanthamoeba sp. is free-living protozoan that is abundant in soil, air, fresh water, and ocean sediments, and is associated with different human diseases, such as cutaneous acanthamoebiasis, lethal granulomatous amoebic encephalitis (GAE), and amoebic keratitis (AK) [
1], and AK is one of the most serious corneal infections. The genus
Acanthamoeba is classified into 17 different genotypes (T1-T17) based on rDNA sequences [
2,
3], although the majority of AK cases are associated with the T4 genotype. At least 9 species of
Acanthamoeba have been identified in ocular infections, that is,
A. griffini (T3),
A. triangularis,
A. castellanii,
A. polyphaga,
A. rhysodes,
A. quina, and
A. lugdunensis (T4),
A. culbertsoni (T10); and
A. hatchetti (T11) [
4-
7].
AK remains a difficult infection to diagnose and manage and is most commonly associated with contact lenses. Recently, the number of young contact lens wearers has increased for correction of myopia and cosmetic purposes [
8,
9], and although rare, AK after corneal laser refractive surgery has been reported [
10,
11]. AK is frequently misdiagnosed initially as herpes simplex virus or bacterial or fungal keratitis, ensuing treatment delays, and often allows progress to a state that may cause significant visual loss or perforating keratopathy requiring corneal transplant surgery [
12,
13]. Therefore, early diagnosis is required to potentiate more favorable prognosis. A diagnosis of AK is usually established by the microscopic identification of
Acanthamoeba from a direct smear of corneal scrapings or cultured specimens, but it is time-consuming and requires technical expertise to discriminate
Acanthamoeba cysts or trophozoites, and unfortunately, this frequently leads to delayed appropriate treatment [
13]. To improve the detection of
Acanthamoeba, molecular diagnostic methods, such as PCR, are increasingly used for its early detection in corneal specimens [
14,
15]. However, although these assays have been shown to be highly effective for diagnosing AK, they require relatively expensive laboratory equipments, and a trained specialist, which limit their usefulnesses in regions where non-contact lens-related AK is prevalent [
16,
17]. Therefore, we considered the need for a simple, rapid, and sensitive diagnostic assay for pathogenic
Acanthamoeba.
Loop-mediated isothermal amplification (LAMP) is a relatively simple, sensitive technique based on rapid DNA amplification [
18]. The technique requires the target DNA to be specifically amplified by
Bst DNA polymerase, which performs strand displacement DNA-synthesis active under isothermal conditions, using a set of 6 oligonucleotides that recognize independent regions of the target gene. The use of 6 oligonucleotides improves the specificity and the speed of amplification and forms a loop-structured amplicon, which produces a typical ladder-pattern of multiple bands [
19]. A positive reaction is easily determined by eye as turbidity [
20] or by adding fluorescent dyes [
21,
22]. LAMP assays have previously been applied to the detection of pathogens, such as viruses, bacteria, fungi, and parasites [
19].
Recently, Lek-Uthai et al. [
23] described a LAMP assay targeting 18S rDNA to detect
Acanthamoeba in contact lens cases. However, the test was not validated with any specified
Acanthamoeba species or clinical ocular samples of AK. Nonetheless, recent increases in the incidence of AK demand an improved diagnostic method. The present study was undertaken to develop a LAMP assay for the detection of
Acanthamoeba in clinical ocular samples to enable the diagnosis of AK. The sensitivity and specificity of the devised assay were determined using specified
Acanthamoeba species and evaluated using clinical ocular samples.
MATERIALS AND METHODS
Samples
The reference
Acanthamoeba species used in this study for specificity and sensitivity experiments were purchased from the American Type Cell Collection (ATCC; Rockville, Maryland, USA) (
Table 1). Amoebae were cultured axenically in peptone-yeast-glucose (PYG) medium at 25℃, as previously described [
24]. Clinical ocular samples (corneal scrapings and contact lens solutions) from suspected keratitis patients that registered at Kyungpook National University Hospital (Daegu, Korea) between March 2011 and March 2013 were assayed using standard diagnostic procedures, such as direct examination and culture. Donor confidentiality was maintained as is required by the Declaration of Helsinki and the study was approved by the Kyungpook National University institutional review board (IRB)/ethics committee (KNUH 2011-04-008).
The sensitivities of LAMP assays were assessed using standard plasmids containing 18S rDNA LAMP targeting fragments (1, 10, 10
2, 10
3, and 10
4 copies) and serially diluted
Acanthamoeba genomic DNA (1, 10, and 100 pg). The 18S rDNA gene was amplified by PCR from each
Acanthamoeba species using primers F3 and B3 (listed in
Table 2). PCR products were purified using a Qiaquick gel extraction kit (Qiagen, Valencia, California, USA). Thereafter, eluted PCR products were cloned into pGEM-T easy vector (Promega, Madison, Wisconsin, USA), and amplified DNA fragments were completely sequenced (Macrogen, Seoul, Korea). Concentrations of plasmid DNA were measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Thermo Scientific Instruments, Wilmington, Delaware, USA) and corresponding copy numbers were calculated. The standard plasmid was diluted with TE buffer (10 mM Tris-HCl, 1 mM EDTA) to final concentrations of 1, 10, 10
2, 10
3, or 10
4 copies of cloned 18S rDNA per microliter. On the other hand, the minimum genomic DNA concentration required for the detection of
Acanthamoeba was determined using serially diluted
Acanthamoeba genomic DNA.
Acanthamoeba trophozoites harvested from cell culture were counted on a hematocytometer and diluted to 1×10
6 trophozoites in 200 µl of PBS. Genomic DNA was extracted using QiaAmp mini kits (Qiagen), quantitated, and serially diluted to 1 µg to 100 fg/µl with TE buffer. DNA from
Aspergillus fumigatus,
Fusarium solani, and
Candida albicans were extracted using recombinant lyticase (Sigma, Deissenhofen, Germany) and a QiaAmp mini kit. DNA of
Entamoeba histolytica and
Giardia lamblia were kindly provided by Dr. Myeong Heon Shin and Dr. Soon-Jung Park, Yonsei University College of Medicine (Seoul, Korea).
Acanthamoeba-specific LAMP primers were designed as previously described by Notomi et al. [
18], and the Primer Explorer program (
http://primerexplorer.jp/elamp4.0.0/index.html; Eiken Chemical Co., Japan) was used to amplify the 18S rDNA gene (
Table 2). The 18S rDNA sequences of
Acanthamoeba sp. used for primer design were retrieved from GenBank or obtained by PCR using F3 and B3 primers for the appropriate genes (
A. castellanii; GenBank no. U07413,
A. triangularis GenBank no. AF316547,
A. rhysodes GenBank no. AY351644,
A. lugdunensis GenBank no. AF005995,
A. polyphaga GenBank no. AF019061,
A. quina GenBank no. AY703023,
A. griffini GenBank no. U07412,
A. hatchetti GenBank no. AF019068,
A. culbertsoni GenBank no. AF019067,
A. healyi GenBank no. AF019070, and
A. astronyxis GenBank no. AF019064). The
Acanthamoeba specific LAMP primer set consisted of F3 (forward outer primer), B3 (backward outer primer), FIP (forward inner primer), BIP (backward inner primer), LF (loop forward primer), and LB (loop backward primer). LAMP was performed for 90 min at 64℃ in a 25 µl mixture containing 40 pmol each of FIP and BIP, 5 pmol each of F3 and B3, 20 pmol each of LF and LB, 1.4 mM of deoxynucleoside triphosphates, 0.8 M betaine, and 1 µl of
Bst DNA polymerase (NEB, Beverly, Massachusetts, USA) in 2.5 µl of buffer (20 mM Tris-HCl pH 8.8, 10 mM KCl, 10 mM (NH
4)
2SO
4, 8 mM MgSO
4, and 0.1% Tween 20), and 1 µl of different concentrations of genomic DNA (100 pg-100 fg) in a Loopamp real-time turbidimeter (Realoop-30; Eiken Chemical, Tokyo, Japan). After LAMP assay, reactions were inactivated for 2 min at 80℃ and then evaluated by electrophoresis using agarose gel (2.0%) or visualized by fluorescence detection reagent (FD; Eiken Chemical) under UV light. To determine the optimal reaction temperature for the 2 LAMP assays, reaction mixtures were incubated for 120 min at 58 to 65℃. The temperature selected was that allowed reaction to exceed the positive reaction threshold turbidity value of 0.1 in the shortest time.
Comparison of LAMP and PCR assays using clinical ocular samples
Clinical samples were suspended in 300 µl of sterile saline and divided into 3 parts. Two parts were used for DNA purification, the other was cultured for
Acanthamoeba detection. DNAs were prepared from clinical ocular samples suspended in 100 µl of sterile saline, as mentioned above, using 2 different methods, that is, either by using a commercially available kit (DNeasy tissue kits, Qiagen) or by heat-treating with NaOH solution. Prior to DNA extraction, specimens were subjected to 3 freeze-thaw cycles using liquid nitrogen and heating (56℃) to disrupt
Acanthamoeba cysts. DNA was extracted using DNeasy tissue kits (Qiagen) in accordance with the manufacturer's instructions. One microliter of extracted DNA dissolved in 20 µl of doubly distilled water was used as the template for the PCR and LAMP assays. On the other hand, heat-treated lysates of clinical samples were prepared as previously described with minor modification [
24]. Briefly, samples were concentrated by centrifugation, resuspended in 20 µl of 0.1N NaOH solution, heated at 100℃ for 3 min to disrupt
Acanthamoeba trophozoites and cysts, and centrifuged at 12,000×g for 2 min at room temperature. One microliter aliquots of the supernatants collected were used as templates for PCR and LAMP.
PCR was performed in triplicate using TaKaRa LA Taq polymerase (Takara Biochemicals, Kyoto, Japan) in a reaction volume of 20 µl containing Ampdirect (Shimadzu, Kyoto, Japan) and a thermal cycler (Perkin Elmer Cetus, Norwalk, Connecticut, USA). The PCR conditions used were as follows: an initiation step of 94℃ for 10 min; 35 cycles of 94℃ for 30 s, 55℃ for 90 s, and 72℃ for 60 s, and a final extension step at 72℃ for 10 min. Two primer sets (Nelson and JDP), specific for multicopy
Acanthamoeba genomic sites encoding rRNA were employed per reaction [
14,
15]. The primer sets had the Nelson sequences (forward, 5'-GTTTGAGGCAATAACAGGT-3'; reverse, 5'-GAATTCCTCGTTGAAGAT-3') and generated a product 229 bp long, and the JPD sequences (forward, 5'-GGCCCAGATCGTTTACCGTGAA-3'; reverse, 5'-TCTCACAAGCTGCTAGGGAGTCA-3') and generated a product 423-551 bp long. PCR products from each reaction were confirmed by DNA sequencing (Macrogen). Amplified products were visualized on 1.5% agarose gels, stained with ethidium bromide, and observed using a UV transilluminator. For culture, specimens concentrated by centrifugation were inoculated onto culture plates containing non-nutrient medium overlaid with heat-treated
E. coli, and then incubated at 25℃ for 14 days. Cultures were examined daily for growth under an inverted microscope. Culture positivity was interpreted as positivity for
Acanthamoeba. The relative sensitivities of PCR tests were calculated by expressing the proportions of positive samples as percentages of the proportions of corresponding culture positive samples. Calculations were made using VassarStats: Website for Statistical Computations (
http://faculty.vassar.edu/lowry/VassarStats.html).
RESULTS
Optimization of Acanthamoeba LAMP assay conditions
To develop a LAMP assay for detecting
Acanthamoeba, we designed a set of 6 LAMP primers targeting the 18S rDNA gene (
Fig. 1), which is highly conserved in
Acanthamoeba sp. The optimal temperature and time for the LAMP reaction were determined using cloned 18S rDNA (10
6 copies per reaction) under isothermal conditions at temperatures of 60 to 65℃ for 120 min, by monitoring turbidity. Although amplification targeting the 18S rDNA gene was detected at all temperatures tested, the LAMP assay targeting the 18S rDNA gene reached a threshold value (0.1) with shortest incubation times at 64℃ (data not shown). No non-specific amplification was detected in the negative control (plasmid containing no insert) after at least 120 min of incubation. Thus, subsequent LAMP reactions were conducted at 64℃ for 90 min.
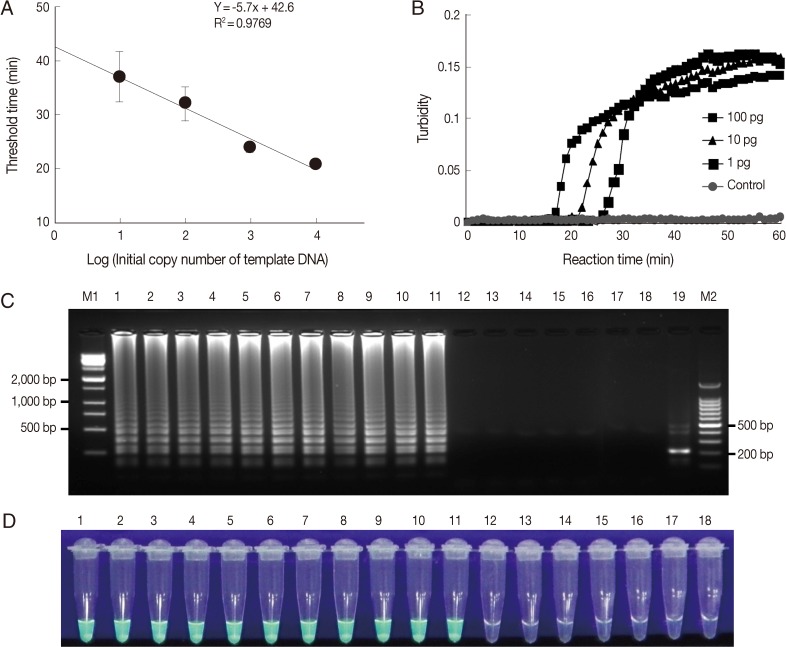
To determine the sensitivity of the LAMP assay, we used plasmid DNA containing the 18S rDNA gene and genomic DNA of each
Acanthamoeba species. First, the LAMP assay was performed using serially diluted plasmid DNA containing the 18S rDNA gene and analyzed by plotting threshold time versus log of the initial template copy number. The detection limit of copy number for a positive turbidity signal was 10 copies of
A. castellanii 18S rDNA per reaction (
Fig. 2A) (linear regression coefficient; 0.9769). In addition, the LAMP reaction was carried out using
Acanthamoeba genomic DNA extracted from in vitro cultured trophozoites. The LAMP procedure amplified serially diluted
A. castellanii genomic DNA at each dilution from the highest DNA concentration examined (100 pg) to as little as 1 pg for 18S rDNA (
Fig. 2B). In 18S rDNA LAMP assays using serially diluted genomic DNA extracted from in vitro cultured
Acanthamoeba species, the minimal genomic DNA concentration for the detection of
Acanthamoeba was 1 pg per reaction for all 11
Acanthamoeba species tested (data not shown). The specificity of the
Acanthamoeba LAMP assay was evaluated using the genomic DNA of 11 known Acanthamoeba species, including species clinically associated with AK (
Table 1), 3 keratitis-related fungal species (
Aspergillus fumigatus,
Fusarium solani, and
Candida albicans), 2 pathogenic, but keratitis-unrelated protozoan parasites (
Entamoeba histolytica and
Giardia lamblia), and 1 bacteria (
Escherichia coli). As shown in
Fig. 2C, a typical ladder pattern of amplified LAMP products on agarose gel electrophoresis was observed for all
Acanthamoeba species tested by 18S rDNA targeting LAMP assays. However, no amplification was detected in LAMP assays on fungal, AK-unrelated protozoan, or bacterial DNAs (
Fig. 2C). Amplified products of positive reactions were also visually detectable using fluorescent detection reagent under UV light (
Fig. 2D). To determine the specificity of the amplification of LAMP assays, amplified LAMP products were digested with
NcoI (
Fig. 1, arrowhead), and this generated the expected 216 bp fragment (
Fig. 2C, lane 19).
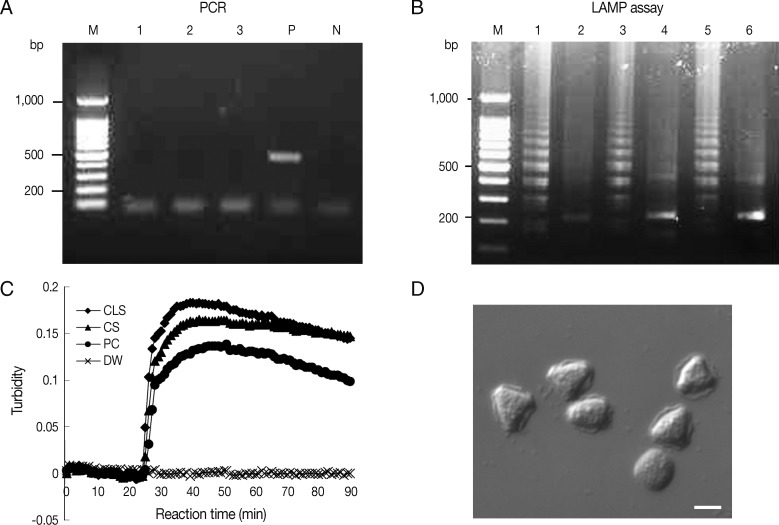
Ocular clinical samples from patients were initially divided into 3 for culture and template DNA preparation for LAMP assays and PCR. In this study,
Acanthamoeba-positivity was defined as culture-positivity. Fifteen
Acanthamoeba-positive ocular clinical samples (11 corneal scrapings and 4 contact lens solutions) from 11 patients diagnosed with
Acanthamoeba infection by microscopy after 14-21 days of in vitro culture, were tested for
Acanthamoeba DNA by LAMP and PCR. Because rapid and simple sample preparations are convenient, inexpensive, and can reduce false-positive results, the template DNAs were prepared from 15 ocular samples using 2 different methods, that is, using a commercial kit (DNeasy tissue kit) or heat-treated specimens in 0.1N NaOH solution. Of these 15 culture positive samples, 9 corneal scrapings and 4 contact lens solutions were positive by PCR using Nelson primers, with sensitivities of 81.8% and 100%, respectively (
Table 3). Results for the amplification of
Acanthamoeba DNA by PCR using JDP primers were positive for 8 corneal scrapings and 4 contact lens solutions, with sensitivities of 72.7% and 100%, respectively (
Table 3). For the LAMP assay, all positive samples by microscopy in culture were positive, that is, the sensitivity of the LAMP assay was 100% for corneal scrapings and contact lens solutions (
Table 3). However, PCR results using direct heat-treated lysates (corneal scrapings and contact lens solutions) showed no amplification of DNA, whereas the LAMP assay using direct heat-treated lysates produced the same results as those obtained using DNA extracted with the commercial kit (
Table 3;
Fig. 3A, B). As shown in
Fig. 3C, which contains representative results, real-time turbidities showed LAMP product amplification within 30 min for heat-treated corneal scrapings and for contact lens solutions lysates collected from a patient, in whom
Acanthamoeba infection was confirmed after 10 days of culture (
Fig. 3D).
DISCUSSION
AK is a vision-threatening disease that is difficult to diagnose and manage, and the incidence of AK is probably increasing, as AK is most commonly associated with contact lens wearing [
1]. Furthermore, effective treatment requires a rapid, sensitive, inexpensive, and easily performed diagnostic method that produces readily interpreted results. Microscopic identification from direct smears or cultured specimens is a highly specific method for detecting
Acanthamoeba, but it is time-consuming and requires technical expertise to discriminate
Acanthamoeba cysts and trophozoites. To solve these problems, new molecular diagnostic tests have been developed. PCR-based methods have been developed to detect
Acanthamoeba in corneal scrapings [
14,
15], but despite the effectivenesses of these assays for
Acanthamoeba detection, relatively expensive laboratory equipment, a trained specialist, and DNA preparation processes are required, which limits their usefulnesses. On the other hand, LAMP can be performed with minimal laboratory facilities, such as a heating block or a water bath. Furthermore, the use of a fluorescent dye or the assessment of turbidity by eyes reduces the need for time-consuming post-PCR procedures, such as agarose gel electrophoresis.
Previous studies have shown that PCR has slightly higher sensitivity (Nelson primer PCR) or lower sensitivity (JDP primer PCR) than the culture of corneal scrapings [
15,
25]. In the present study, Nelson primer PCR was more effective at DNA amplification than JDP primer PCR, which is consistent with previous reports [
25]. Using culture positivity as the "gold standard", the sensitivities of Nelson primer PCR and JDP primer PCR were lower than that of LAMP assay, indicating that amplification of
Acanthamoeba DNA by LAMP is more sensitive for the diagnosis of AK. Furthermore, straightforward sample preparation reduces the risk of cross-contamination, and thus, the risks posed by false-positive results. In the present study, LAMP assay based on directly heat-treated samples was found to be as efficient at detecting
Acanthamoeba as DNA extracted using a commercial kit, whereas PCR was only effective when kit-extracted DNA was used (
Table 3). This difference between LAMP and PCR results is probably due to the ability of LAMP to better tolerate biological contaminants [
26,
27]. Thus, the devised
Acanthamoeba LAMP assay could eliminate the need for DNA extraction without compromising sensitivity and reduce the time required to reach a diagnosis. Furthermore, the simplicity, rapid sample preparation, and straightforward detection of LAMP suggest its potential applications in regions where non-contact lens-related AK is prevalent [
16,
17]. Although, in the present study the LAMP assay was only applied to corneal ocular specimens, the assay could be used to detect GAE-causing
Acanthamoeba in immunocompromised hosts. GAE presents as a subacute but progressive meningoencephalitis that is almost universally fatal [
28], and thus, we consider it worth investigating whether this assay could be modified to enable the diagnoses of amoebic infections using cerebrospinal fluid or brain tissue samples. Furthermore,
Acanthamoeba is one of the most common protists in soil and is also found in freshwater, cooling towers, and sewage systems [
1], and thus, the described LAMP assay could be used for the active surveillance of
Acanthamoeba.
Recently, several studies have described LAMP assays targeting 18S rDNA for the detection of
Acanthamoeba in contact lens cases [
23]. However, the effectivenesses of these tests were not evaluated on
Acanthamoeba sp. from ATCC reference strains, with the exception of
A. castellanii. Moreover, the LAMP amplicon shows a typical ladder-pattern of multiple bands, as shown in
Fig. 3A, but only a single broad band of LAMP-amplified products by gel electrophoresis [
23]. Although our LAMP assay also targeted the
Acanthamoeba 18S rDNA gene, we designed our primers based on multiple DNA sequence alignments that can detect most
Acanthamoeba sp, including all known AK-associated species. Furthermore, 2 additional loop primers were included to enhance the amplification reaction and to reduce reaction times.
Summarizing, we describe the development and validation of a LAMP assay for the detection of Acanthamoeba DNA from clinical ocular samples, and suggest that this technique be used to supplement traditional culture for the detection of AK, especially when diagnostic equipment is minimal.
ACKNOWLEDGMENTS
This study was supported by a grant of the Korea Healthcare Technology R&D Project, Ministry of Health & Welfare (Grant no. A100472). We are indebted to Dr. Soon-Jung Park and Myeong Heon Shin (Yonsei University College of Medicine, Seoul, Korea) for providing the E. histolytica and G. lamblia DNA and to the Korean Collection of Medical Fungi for providing A. fumigatus, F. solani, and C. albicans DNA. We also thank the National Research Center for Protozoan Diseases and the Obihiro University of Agriculture and Veterinary Medicine (Obihiro, Japan).
References
- 1. Khan NA. Acanthamoeba: biology and increasing importance in human health. FEMS Microbiol Rev 2006;30:564-595.
- 2. Corsaro D, Venditti D. Phylogenetic evidence for a new genotype of Acanthamoeba (Amoebozoa, Acanthamoebida). Parasitol Res 2010;107:233-238.
- 3. Nuprasert W, Putaporntip C, Pariyakanok L, Jongwutiwes S. Identification of a novel T17 genotype of Acanthamoeba from environmental isolates and T10 genotype causing keratitis in Thailand. J Clin Microbiol 2010;48:4636-4640.
- 4. Ledee DR, Hay J, Byers TJ, Seal DV, Kirkness CM. Acanthamoeba griffini. Molecular characterization of a new corneal pathogen. Invest Ophthalmol Vis Sci 1996;37:544-550.
- 5. Walochnik J, Obwaller A, Aspock H. Correlations between morphological, molecular biological, and physiological characteristics in clinical and nonclinical isolates of Acanthamoeba spp. Appl Environ Microbiol 2000;66:4408-4413.
- 6. Yu HS, Kong HH, Kim SY, Hahn YH, Hahn TW, Chung DI. Laboratory investigation of Acanthamoeba lugdunensis from patients with keratitis. Invest Ophthalmol Vis Sci 2004;45:1418-1426.
- 7. Xuan YH, Chung BS, Hong YC, Kong HH, Hahn TW, Chung DI. Keratitis by Acanthamoeba triangularis: report of cases and characterization of isolates. Korean J Parasitol 2008;46:157-164.
- 8. Dart JK, Saw VP, Kilvington S. Acanthamoeba keratitis: diagnosis and treatment update 2009. Am J Ophthalmol 2009;148:487-499.e2.
- 9. Patel A, Hammersmith K. Contact lens-related microbial keratitis: recent outbreaks. Curr Opin Ophthalmol 2008;19:302-306.
- 10. Mozayan A, Madu A, Channa P. Laser in-situ keratomileusis infection: review and update of current practices. Curr Opin Ophthalmol 2011;22:233-237.
- 11. Garg P, Chaurasia S, Vaddavalli PK, Muralidhar R, Mittal V, Gopinathan U. Microbial keratitis after LASIK. J Refract Surg 2010;26:209-216.
- 12. Panjwani N. Pathogenesis of Acanthamoeba keratitis. Ocul Surf 2010;8:70-79.
- 13. Hammersmith KM. Diagnosis and management of Acanthamoeba keratitis. Curr Opin Ophthalmol 2006;17:327-331.
- 14. Mathers WD, Nelson SE, Lane JL, Wilson ME, Allen RC, Folberg R. Confirmation of confocal microscopy diagnosis of Acanthamoeba keratitis using polymerase chain reaction analysis. Arch Ophthalmol 2000;118:178-183.
- 15. Schroeder JM, Booton GC, Hay J, Niszl IA, Seal DV, Markus MB, Fuerst PA, Byers TJ. Use of subgenic 18S ribosomal DNA PCR and sequencing for genus and genotype identification of acanthamoebae from humans with keratitis and from sewage sludge. J Clin Microbiol 2001;39:1903-1911.
- 16. Sharma S, Srinivasan M, George C. Acanthamoeba keratitis in non-contact lens wearers. Arch Ophthalmol 1990;108:676-678.
- 17. Srinivasan M, Burman S, George C, Nirmalan PK. Non-contact lens related Acanthamoeba keratitis at a tertiary eye care center in south India: Implications for eye care programs in the region. Med Sci Monit 2003;9:CR125-CR129.
- 18. Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 2000;28:E63.
- 19. Mori Y, Notomi T. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J Infect Chemother 2009;15:62-69.
- 20. Mori Y, Nagamine K, Tomita N, Notomi T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem Biophys Res Commun 2001;289:150-154.
- 21. Goto M, Honda E, Ogura A, Nomoto A, Hanaki K. Colorimetric detection of loop-mediated isothermal amplification reaction by using hydroxy naphthol blue. Biotechniques 2009;46:167-172.
- 22. Tomita N, Mori Y, Kanda H, Notomi T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat Protoc 2008;3:877-882.
- 23. Lek-Uthai U, Passara R, Roongruangchai K, Buddhirakkul P, Thammapalerd N. Rapid identification of Acanthamoeba from contact lens case using loop-mediated isothermal amplification method. Exp Parasitol 2009;121:342-345.
- 24. Kong HH, Shin JY, Yu HS, Kim J, Hahn TW, Hahn YH, Chung DI. Mitochondrial DNA restriction fragment length polymorphism (RFLP) and 18S small-subunit ribosomal DNA PCR-RFLP analyses of Acanthamoeba isolated from contact lens storage cases of residents in southwestern Korea. J Clin Microbiol 2002;40:1199-1206.
- 25. Boggild AK, Martin DS, Lee TY, Yu B, Low DE. Laboratory diagnosis of amoebic keratitis: comparison of 4 diagnostic methods for different types of clinical specimens. J Clin Microbiol 2009;47:1314-1318.
- 26. Enomoto Y, Yoshikawa T, Ihira M, Akimoto S, Miyake F, Usui C, Suga S, Suzuki K, Kawana T, Nishiyama Y, Asano Y. Rapid diagnosis of herpes simplex virus infection by a loop-mediated isothermal amplification method. J Clin Microbiol 2005;43:951-955.
- 27. Kaneko H, Kawana T, Fukushima E, Suzutani T. Tolerance of loop-mediated isothermal amplification to a culture medium and biological substances. J Biochem Biophys Methods 2007;70:499-501.
- 28. Booton GC, Visvesvara GS, Byers TJ, Kelly DJ, Fuerst PA. Identification and distribution of Acanthamoeba species genotypes associated with nonkeratitis infections. J Clin Microbiol 2005;43:1689-1693.
Fig. 1Nucleotide sequence alignments of target regions in the Acanthamoeba 18S rDNA genes of the 11 tested Acanthamoeba species. Primer recognition sites are indicated by arrows and gray boxes with primer names. Black boxes indicate non-conserved nucleotides. The black arrowhead indicates the NcoI cleavage site. F3, forward outer primer; B3, backward outer primer; FIP, forward inner primer; BIP, backward inner primer; LF, loop forward primer; and LB, loop backward primer.

Fig. 2Sensitivities and specificities of Acanthamoeba LAMP assays. LAMP assays were performed using serial dilutions of (A) plasmid DNA containing Acanthamoeba castellanii 18S rDNA (10, 102, 103, or 104 copies per reaction) and (B) genomic DNA (100, 10, or 1 pg). Plasmid containing no insert was used as a control. LAMP products were visualized by (C) gel electrophoresis and using (D) the Loopamp® fluorescent detection reagent (FD). Lanes M1 and M2, 1-kb and 100-bp molecular weight markers, respectively; lane 1, Acanthamoeba astronyxis; lane 2, Acanthamoeba triangularis; lane 3, Acanthamoeba rhysodes; lane 4, Acanthamoeba castellanii; lane 5, Acanthamoeba lugdunensis; lane 6, Acanthamoeba polyphaga; lane 7, Acanthamoeba quina; lane 8, Acanthamoeba griffini; lane 9, Acanthamoeba hatchetti; lane 10, Acanthamoeba culbertsoni; lane 11, Acanthamoeba healyi; lane 12, Aspergillus fumigatus; lane 13, Fusarium solani; lane 14, Candida albicans; lane 15, Entamoeba histolytica; lane 16, Giardia lamblia; lane 17, Escherichia coli; lane 18, distilled water; and lane 18, NcoI digestion of the LAMP product of 18S rDNA.

Fig. 3Detection of Acanthamoeba in heat-treated ocular clinical samples by LAMP and PCR; (A and B) Electrophoresis of PCR (A) and LAMP assay products (B) amplified from heat-treated ocular clinical samples from a patient with suspected keratitis. (A) Lanes 1-2, heat-treated lysates of contact lens solutions from left and right lens case chambers, respectively; lane 3, heat treated lysate of corneal scrapings; lane P, 1 pg of A. castellanii genomic DNA (positive control); lane N, negative control (distilled water); lane M, 100-bp ladder DNA marker. (B) Lane 1, heat-treated lysate of contact lens solutions from the right lens case chamber; lane 3, heat-treated lysate of corneal scrapings; lane 5, 1 pg of A. castellanii genomic DNA (positive control); lanes 2, 4, and 6; NcoI digestions of the LAMP products of lanes 1, 3, and 5, respectively. (C) Real-time turbidity LAMP assay of the sample in (B). CLS, contact lens solution; CS, corneal scraping; PC, positive control; DW, distilled water. (D) Photographs of trophozoites of Acanthamoeba sp. detected by cultures of corneal scrapings and a contact lens solution of the same samples mentioned in (B) (original magnification×400, scale bar=10 µm).

Table 1.List of Acanthamoeba reference species used in the LAMP assay
Table 1.
|
Species |
Genotype |
Strain |
ATCC No. |
Source |
Geographic source |
|
Acanthamoeba griffini
|
T3 |
S-7 |
30731 |
Beach-bottom |
United States |
|
Acanthamoeba triangularis
|
T4 |
SH621 |
50254 |
Human feces |
France |
|
Acanthamoeba rhysodes
|
T4 |
Singh |
30973 |
Soil |
England |
|
Acanthamoeba castellanii
|
T4 |
Castellani |
30011 |
Yeast culture |
England |
|
Acanthamoeba lugdunensis
|
T4 |
L3a |
50240 |
Swimming pool |
France |
|
Acanthamoeba polypaga
|
T4 |
Page23 |
30871 |
Freshwater |
United States |
|
Acanthamoeba quina
|
T4 |
Vil3 |
50241 |
Swimming pool |
France |
|
Acanthamoeba astronyxis
|
T7 |
Ray & Hayes |
30137 |
Soil |
United States |
|
Acanthamoeba culbertsoni
|
T10 |
Lilly A-1 |
30171 |
Tissue culture |
United States |
|
Acanthamoeba hatchetti
|
T11 |
BH-2 |
30730 |
Ocean sediment |
United States |
|
Acanthamoeba healyi
|
T12 |
OC-3A |
30866 |
GAEa
|
United States |
Table 2.The sequences of the LAMP primer sets used to amplify Acanthamoeba 18S rDNA genes for the LAMP assay
Table 2.
|
Target gene |
Primer |
Sequence (5´→3´) |
|
18S rDNA |
F3 |
GGCGACGATTCATTCAAAT |
|
B3 |
CAAGACTCTTGTCGAGCGC |
|
FIP |
TCCCTCTCCGGAATCGAACCCTCGATGGTAGGATAGAGGCC |
|
BIP |
TTCTAAGGAAGGCAGCAGGCGTATTGTCACTACCTCCCCGT |
|
LF |
TCCGTTACCCGTTACGACCA |
|
LB |
CGCAAATTACCCAATCCCGAC |
Table 3.Detection of Acanthamoeba by the conventional PCR and LAMP assay using 2 differently prepared samples
Table 3.
|
Assay |
No. of samples with the following culture detection methods (%)
|
Commercial kit
|
Heat treated/NaOH
|
|
Corneal scrapings (n = 11) |
Contact lens sdutions (n = 4) |
Corneal scrapings (n = 11) |
Contact lens sdutions (n=4) |
|
18S rDNA PCR |
|
|
|
|
|
Nelson primers |
9 (81.8) |
4 (100.0) |
0 |
0 |
|
JDP primers |
8 (72.7) |
4 (100.0) |
0 |
0 |
|
18S rDNA LAMP |
11 (100) |
4 (100.0) |
11 (100.0) |
4 (100.0) |