Abstract
There have been few reports on extra-enteric infections by Blastocystis STs and none have been molecularly identified in samples from human reproductive organs. We report for the first time the identification of 3 different subtypes of Blastocystis (ST1–3) in vaginal and sperm samples, from patients infected with Trichomonas vaginalis. Blastocystis STs were identified by PCR-sequencing and by phylogenetic inferences using 28 vaginal swab samples and 7 sperm samples from patients trichomoniasis. Blastocystis STs were identified in 6 of 28 vaginal swabs (21.4%) and in 3 of 7 sperm samples (42.8%). In both biological samples, STs 1–3 were found; one vaginal sample showed subtype co-infection with ST1 and ST3. High genetic variation was observed in the sequences obtained and no specific clustering in the phylogenetic trees was detected. Most of the haplotypes identified were placed far from the main dispersal centers. Our finding suggested that incorrect cleaning of the genital area or a contamination by combination of anal and vaginal intercourse.
-
Key words: Blastocystis subtypes, Trichomonas vaginalis, extra-enteric blastocystosis
Blastocystis is among the most common intestinal microeukaryote identified in stool samples worldwide. Its pathogenic role remains controversial; several studies have linked this microbe to the development of dermatological and intestinal disorders, while others have not.
Blastocystis exhibits a great variability in the size and shape of its main stages observed under the light microscope (granular and vacuolar forms). Furthermore, it displays a marked genetic polymorphism. Up to 28 ribosomal lineages proposed, known as subtypes (STs), have been described by analyzing the small subunit 18S ribosomal RNA (18S rRNA) [
1].
There are few cases of extra-enteric
Blastocystis in humans, such as those found in knee synovial fluid [
2], liver, and splenic abscess [
3–
5]. A previous study examined vaginal parasites in 312 Polish peasant women and found 47 persons infected with
Blastocystis (11.5%) [
6]. A case of
Blastocystis in the uterine cervix of a Mexican patient with minimal cervicitis symptoms was recently reported [
7]. Interestingly, in all cases, the vacuolar stage was clearly identified by microscopy but molecular diagnosis of STs has not been conducted.
On the other hand, the protozoan
Trichomonas vaginalis is the most common nonviral sexually transmitted infection (STI) in the world.
T. vaginalis inhabits the vagina, causing vaginitis and cervicitis. In male it colonizes the urethra, epididymis, and prostate gland, but usually asymptomatic. More importantly, however,
Trichomonas infections can cause infertility in both men and women [
8].
Identification of Blastocystis in the cervix of a Mexican patient who was coinfected with T. vaginalis led us to investigate the Blastocystis STs in these samples. In the present study, we report the unexpected identification of Blastocystis STs in human vaginal and sperm samples coinfected with T. vaginalis.
Twenty-eight women who attended the gynecology public service at the Hospital General Dr. Manuel Gea Gonzalez (HGMGG) for routine check-ups were analyzed. The indications for sampling included i) not having ingested any antibiotics for at least 10 days prior to medical appointment, ii) without the application of ovules or vaginal creams, iii) not having had sexual intercourse for at least 3 days, iv) not menstruating or with abnormal bleeding, and iv) properly cleaned. The samples of vaginal swabs were put in containers BD Vacutainer with sterile physiological saline and diagnosed with
T. vaginalis by microscopy. Immediately after diagnosis, the samples were kept at −70°C. In men, aliquots of 7 sperm samples that were employed for spermatobioscopy or assisted reproduction treatment were donated from the private fertility clinic of Biolab, Andrology laboratory. The indications for semen sampling included i) not having had sexual intercourse for at least 3 days, ii) not having taken any antibiotics for at least 10 days, and iii) properly cleaned. The samples were obtained in sterile plastic containers of 50 ml and analyzed as recommended by the World Health Organization [
9]. These samples were diagnosed by microscopy with
T. vaginalis, all patients were asymptomatic; the samples were frozen at −70°C.
As part of a routine process, clinical and microbiology data were obtained from medical records of each woman. We also requested diagnosis for other STI such as Gardnerella vaginalis, Ureaplasma urealyticum, Micoplasma hominis, and Chlamydia spp., as well as microbiological and mycological cultures. All samples were collected from 2015 to 2019.
Ethics and Research Committees of the HGMGG approved the study with the reference number 12-75-2015. Written informed consent was obtained from all patients.
Total DNA was extracted from approximately 500 μl of the physiological saline immersed with vaginal swabs or 100 μl of sperm samples using Puregene DNA Purification System (Gentra Systems, Germany) according to the manufacturer’s protocol. To confirm the presence of
T. vaginalis in vaginal and sperm samples, internal transcribed spacer 1 (ITS1)-5.8 S rRNA-ITS2 region was amplified using primers described previously [
10]. Additionally, the presence of
Blastocystis ST [
11] was searched by PCR-sequencing. To rule out STs co-infections, PCR using ST-specific primers (for ST1, ST2, and ST3) were performed: Primers SB83 F 5′-GAAGGACTCTCTGACGATGA-3′ and R 5′-GTCCAAATGAAAGGCAGC-3′ for ST1, yielding an amplicon of 351bp; SB340 F 5′-TGTTCTTGTGTCTTCTCAGCTC-3′ and R 5′-TTCTTTCACACTCCCGTCAT-3′ for ST2, yielding an amplicon of 704 bp; SB227 F 5′-TAGGATTTGGTGTTTGGAGA-3′ and R 5′-TTAGAAGTGAAGGAGATGGAAG-3′ for ST3, yielding an amplicon of 526 bp [
12,
13]. Samples with mixed STs infections were submitted to sequencing 3 more times to obtain the sequences for both STs.
The PCR products were purified and sequenced in both directions. All sequences were subjected to a BLAST search in the GenBank database (MZ700049–MZ700058). Multiple alignments were performed using the MUSCLE tool from the MEGA Program. The algorithms used to obtain the phylogenetic analyses were Neighbor Joining (NJ), maximum likelihood (ML), and Bayesian algorithms (BA). NJ and ML were performed with 1,000 bootstrap replicas under the Tamura-Nei model of evolution, and the algorithms were implemented in MEGA7. For the BA reconstruction, the General Time Reversible model of evolution specifying a gamma distribution and invariable sites (GTR+G+I) was used. Mr. Bayes version 3.4 was used [
14], and the analysis was performed with 5×10
6 generations, sampling trees every 100 generations. Trees with a score lower than those at the stationary phase (burn-in) were discarded, and trees that reached the stationary phase were collected and used to build consensus trees. Sequences of
Blastocystis spp. (isolates representing ST1–ST14) were taken from the GenBank and incorporated into the phylogenetic analysis. A Median Joining Network analysis was performed using NETWORK 4.611 with 906 sequences (
Supplementary Table S1) obtained from the GenBank, which showed high identity with the samples obtained in the present study according to the BLAST search, haplotype networks were established by default settings and assumptions.
Descriptive statistics are expressed as mean and standard deviation (SD). Analysis by Mantel-Haenszel test was performed to find associations. Data analysis was performed with SSPS software Version 15.0 (SPSS Institute, Chicago, Illinois, USA).
The mean female age was 34 (SD=13.7) years and for men it was 32 (SD=2.9) years; all patients lived in Mexico City.
Table 1 summarizes the main clinical and microbiological characteristics of the patients whose samples were analyzed in this study. Amplicons corresponding to
Blastocystis STs were identified in 6 out of 28 vaginal samples (21.4%) and in 3 out of 7 sperm samples (42.8%) (
Supplementary Fig. S1). In both biological samples STs 1–3 were found; interestingly, 1 vaginal sample showed a mixed infection with ST1 and ST3 (
Table 2). No statistical associations between variables were found.
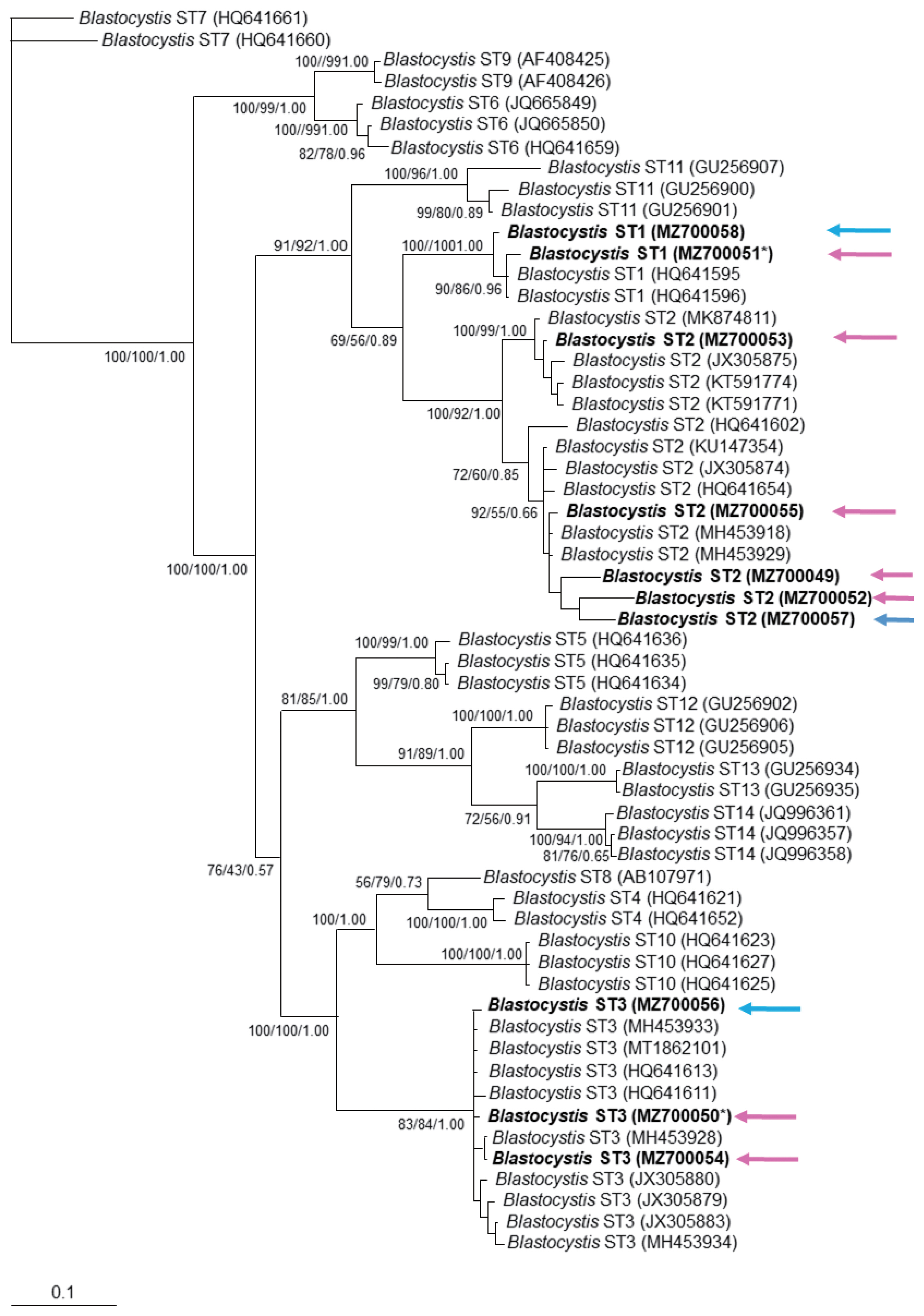
The phylogenetic tree supported the grouping for found sequences inside ST1–3 clusters (
Fig. 1). Similarly, the haplotype network tree grouped sequences in each ST according to other Mexican haplotypes and no specific clustering for the vaginal or sperm samples was observed. Interestingly, most haplotypes identified in the present study were placed far from the main dispersal centers, as “isolated haplotypes” due to some mutations that moved them away from their closest haplotype (
Fig. 2).
To our knowledge, this is the first report of unusual
Blastocystis ST1–3. The presence of
Blastocystis DNA in vaginal and sperm samples is relevant, suggesting that this microorganism has great plasticity in colonizing niches, unlike to other report [
7], where
Blastocystis was only observed morphologically in a vaginal sample by Papanicolaou staining and parasite presence was not clearly identified.
Interestingly, all the samples analyzed for
Blastocystis were
Trichomonas positive by microscopy. The associations between infection by
T. vaginalis and other STIs, particularly of viral infections such as HIV, HSV-2, and HVP have been described [
15]. These studies have shown that there is a high risk of acquiring a viral infection in the women infected with
T. vaginalis. The susceptibility to a viral infection could be increased due to the following reasons: i) the inflammatory response to
Trichomonas infection can promote a greater appearance of target cells; ii)
Trichomonas can damage the mechanical barrier against the virus, and iii) change the normal vaginal microbiota making it more permissive for bacterial vaginosis [
16]. The presence of
T. vaginalis could also increase the susceptibility to
Blastocystis infection. Future prospective cohort study is warranted to identify possible associations between the coinfection of
Trichomonas and
Blastocystis, in addition to seek other potential risk factors.
Previous studies assumed that the women acquired
Blastocystis by anal-genital transmission during incorrect cleaning of the genital area or by combining anal and vaginal intercourse [
6,
7]. This statement is strengthened because
Enterococcus faecalis was documented in her medical record. Since sperm samples can be infected in the same manner,
Blastocysts are likely to be present in the male reproductive system. Among the sexually transmitted parasitic protozoa, only 5 species have been demonstrated in human seminal fluid samples, which included
Entamoeba histolytica,
Schistosoma haematobium,
Trichomonas vaginalis,
Trypanosoma cruzi, and
Toxoplasma gondii [
17]. Furthermore, the presence of
E. histolytica in human seminal fluid was reported once in 1923 in a 28-year-old patient who had chronic amebic dysentery with a history of STIs [
18]. We speculate that high-risk sexual activity related to STIs could increase susceptibility to acquire
Blastocystis. Further elucidation by appropriate causal study design in the future awaits.
The sequences obtained here were clearly grouped into clusters ST1, ST2, and ST3, without a specific distribution according to host’s symptomatology, which is similar to other molecular phylogenetic trees from symptomatic and asymptomatic
Blastocystis carriers [
13]. There was only a vaginal sample with a mixed infection with ST1 and ST3. This finding is consistent with a previous study reporting mixed infections of ST1 and other ST (approximately 10%) [
19].
Blastocystis ST3 was found in splenic cysts [
5], but our study showed that ST1 and ST2 genotypes were also found in extra-intestinal site.
An interesting distribution in the network tree was observed. Most haplotypes identified in this study is located far from the main dispersal centers and appear to “isolate haplotypes”. Some mutations have driven them away from their closest haplotypes that contain great genetic diversity. It has been argued that
Blastocystis can infect several host species and colonize new environments (generalist parasites), this capacity may be the key process for maintaining their genetic diversity and population diversification to parasite some particular species or niches [
20].
This study included several limitations. First, fecal samples were not collected from participants to microscopically identify Blastocystis or compare the genomes of intestinal and extraintestinal parasites from the same host. However, it is worth reporting that T. vaginalis-infected sperm and vaginal samples were simultaneously infected with the Blastocystis ST 1–3 haplotype. Second, we speculate that it is an anal-genital transmission, either during incorrect cleaning of the genital area or during combining anal and vaginal intercourse because the same Blastocystis haplotypes are identified in intestinal and vaginal samples. The possibility of a colonization process of new niches by this microorganism should be tested in future studies.
Notes
-
The authors declare no conflict of interest
Supplementary Information
ACKNOWLEDGMENTS
To patients who provided samples and authorities of the General Hospital Dr. Manuel Gea Gonzalez.
Fig. 1Phylogenetic inference of 18S rRNA gene partial sequence of Blastocystis STs. The values of the nodes indicate the bootstrap proportions and Bayesian posterior probabilities in the following order: Neighbor Joining/maximum likelihood/Bayesian analysis. GenBank accession number are shown at the end of each branch in bold letters. Pink arrows show sequences obtained from vaginal samples, while blue arrows show those for sperm samples. *indicates the sample with mixed Blastocystis STs infection.

Fig. 2Haplotype networks for Blastocystis ST1–3 using 18S rRNA sequences from different countries for ST1 to ST3. Numbers in branches refer to mutational changes; sizes of circles and colors are proportional to haplotype frequencies. Arrows show the haplotypes identified in the present study. Pink arrows show sequences obtained from vaginal samples, while blue arrows show those for sperm samples. *indicates the sample with mixed Blastocystis ST infections.
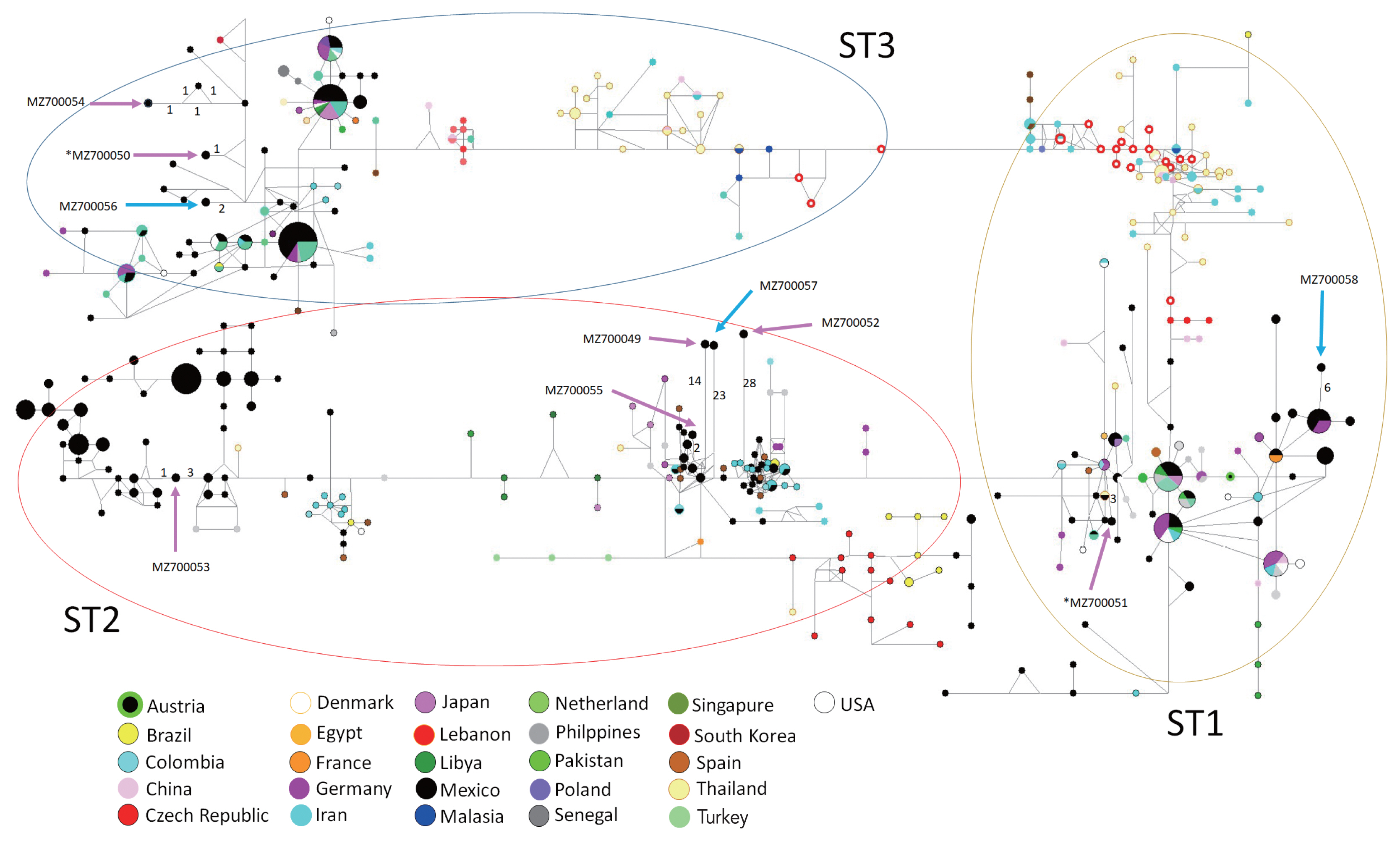
Table 1Main clinical status and microbiological co-infections identified in 28 vaginal samples from symptomatic women and 7 asymptomatic men
Table 1
|
Status |
No. of positives/Total No. of samples (%) |
No. of positives to Blastocystis
|
|
Pregnancy |
12/28 (42.8)*
|
3*
|
|
Overweight/obesity |
13 [9 female+4 male]/35 (37.1) |
2 [both female] |
|
Gestational diabetes |
5/28 (17.8)*
|
2*
|
|
Co-infections |
|
Gardnerella vaginalis
|
9/28 (32.1)*
|
3*
|
|
Ureaplasma urealyticum
|
11/28 (39.3)*
|
3*
|
|
Micoplasma hominis
|
7/28 (25)*
|
2*
|
|
Enterococcus faecalis
|
1/28 (3.6)*
|
0 |
|
Trichomonas vaginalis
|
35/35 (100) |
9 |
|
Chlamydia
|
0 |
0 |
Table 2No. of Blastocystis STs identified in vaginal and sperm samples
Table 2
|
Blastocystis identification |
No. of positives/Total No.of samples (%) |
GenBank access |
|
In vaginal samples |
|
ST2 |
4/28 (14.2) |
MZ700049, MZ700052-3, MZ700055 |
|
ST3 |
1/28 (3.6) |
MZ700054 |
|
ST1+ST3 (coinfections) |
1/28 (3.6) |
MZ700050-1 |
|
|
In sperm samples |
|
ST1 |
1/7 (14.3) |
MZ700058 |
|
ST2 |
1/7 (14.3) |
MZ700057 |
|
ST3 |
1/7 (14.3) |
MZ700056 |
References
- 1. Poppruk S, Adao DEV, Rivera WL. Epidemiology and subtype distribution of Blastocystis in humans: a review. Infect Genet Evol 2021;95:105085. https://doi.org/10.1016/j.meegid.2021.105085
- 2. Lee MG, Rawlins SC, Didier M, De Ceulaer K. Infective arthritis due to Blastocystis hominis. Ann Rheum Dis 1990;49:192-193. https://doi.org/10.1136/ard.49.3.192
- 3. Hu KC, Lin CC, Wang TE, Liu CY, Chen MJ, Chang WH. Amoebic liver abscess or is it? Gut 2008;57:627. https://doi.org/10.1136/gut.2006.119149
- 4. Prodeus TV, Zeila OP, Khlebnikova TA, Pikul’ DA. Extraenteric infection caused by Blastocystis spp. in a female patient with liver abscess. Med Parazitol (Mosk) 2014;2:6-9. (in Russian)..
- 5. Santos HL, Sodré FC, de Macedo HW. Blastocystis sp. in splenic cysts: causative agent or accidental association? A unique case report. Parasit Vectors 2014;7:207. https://doi.org/10.1186/1756-3305-7-207
- 6. Wolyńska M, Soroczan W. Blastocystis hominis Brumpt, 1912, (Phycomycetes) in the female genital tract. Pol Tyg Lek 1972;27:788-791. (in Polish)..
- 7. Escutia-Guzman Y, Martinez-Flores WA, Martinez-Ocaña J, Martinez-Pimentel R, Benitez-Ramirez M, Martinez-Hernandez F, Arroyo-Escalante S, Romero-Valdovinos M, Orozco-Mosqueda GE, Maravilla P. An unusual case of extra-enteric Blastocystosis in the uterine cervix. Korean J Parasitol 2020;58:571-576. https://doi.org/10.3347/kjp.2020.58.5.571
- 8. Shiadeh MN, Niyyati M, Fallahi S, Rostami A. Human parasitic protozoan infection to infertility: a systematic review. Parasitol Res 2016;115:469-477. https://doi.org/10.1007/s00436-015-4827-y
- 9. World Health Organization. WHO Laboratory Manual for the Examination and Processing of Human Semen. 6th ed. World Health Organization; Geneva, Switzerland. 2021. p. 68-69.
- 10. Lopez-Escamilla E, Sanchez-Aguillon F, Alatorre-Fernandez CP, Aguilar-Zapata D, Arroyo-Escalante S, Arellano T, Moncada-Barron D, Romero-Valdovinos M, Martinez-Hernandez F, Rodriguez-Zulueta P, Maravilla P. New Tetratrichomonas species in two patients with pleural empiema. J Clin Microbiol 2013;51:3143-3146. https://doi.org/10.1128/JCM.01056-13
- 11. Santín M, Gómez-Muñoz MT, Solano-Aguilar G, Fayer R. Development of a new PCR protocol to detect and subtype Blastocystis spp. from humans and animals. Parasitol Res 2011;109:205-212. https://doi.org/10.1007/s00436-010-2244-9
- 12. Yoshikawa H, Wu Z, Nagano I, Takahashi Y. Molecular comparative studies among Blastocystis isolates obtained from humans and animals. J Parasitol 2003;89:585-594. https://doi.org/10.1645/0022-3395(2003)089[0585:MCSABI]2.0.CO;2
- 13. Gonzalez-Arenas NR, Villalobos G, Vargas-Sanchez GB, Avalos-Galarza CA, Marquez-Valdelamar LM, Ramirez-Miranda ME, Olivo-Diaz A, Romero-Valdovinos M, Martinez-Hernandez F, Maravilla P. Is the genetic variability of Cathepsin B important in the pathogenesis of Blastocystis spp.? Parasitol Res 2018;117:3935-3943. https://doi.org/10.1007/s00436-018-6103-4
- 14. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003;19:1572-1574. https://doi.org/10.1093/bioinformatics/btg180
- 15. Kissinger P. Trichomonas vaginalis: a review of epidemiologic, clinical and treatment issues. BMC Infect Dis 2015;15:307. https://doi.org/10.1186/s12879-015-1055-0
- 16. Moodley P, Connolly C, Sturm AW. Interrelationships among human immunodeficiency virus type 1 infection, bacterial vaginosis, trichomoniasis, and the presence of yeasts. J Infect Dis 2002;185:69-73. https://doi.org/10.1086/338027
- 17. Crespillo-Andujar C, Díaz-Menéndez M, Mora-Rillo M. Evidence for previously unidentified sexual transmission of protozoan parasites. Emerg Infect Dis 2018;24:602-603. https://doi.org/10.3201/eid2403.171838
- 18. Hines LA. Entamoeba histolytica in seminal fluid in a case of amebic dysentery. JAMA 1923;81:274-275. https://doi.org/10.1001/jama.1923.02650040014003
- 19. Pandey PK, Verma P, Marathe N, Shetty S, Bavdekar A, Patole MS, Stensvold CR, Shouche YS. Prevalence and subtype analysis of Blastocystis in healthy Indian individuals. Infect Genet Evol 2015;31:296-299. https://doi.org/10.1016/j.meegid.2015.02.012
- 20. Villanueva-Garcia C, Gordillo-Chavez EJ, Lopez-Escamilla E, Rendon-Franco E, Muñoz-Garcia CI, Gama L, Martinez-Flores WA, Gonzalez-Rodriguez N, Romero-Valdovinos M, Diaz-Lopez H, Galian J, Villalobos G, Maravilla P, Martinez-Hernandez F. Clarifying the cryptic host specificity of Blastocystis spp. isolates from Alouatta palliata and A. pigra howler monkeys. PLoS One 2017;12:e0169637. https://doi.org/10.1371/journal.pone.0169637

 , Fabiola Sanchez-Aguillon1, Marcia Veronica Carmona-Maldonado2, Nelly Raquel Gonzalez-Arenas1, Eduardo Lopez-Escamilla1, Rigoberto Hernandez-Castro1, Mirza Romero-Valdovinos1, Williams Arony Martinez-Flores1, Juan Pablo Ramirez-Hinojosa1, Pablo Maravilla1,*
, Fabiola Sanchez-Aguillon1, Marcia Veronica Carmona-Maldonado2, Nelly Raquel Gonzalez-Arenas1, Eduardo Lopez-Escamilla1, Rigoberto Hernandez-Castro1, Mirza Romero-Valdovinos1, Williams Arony Martinez-Flores1, Juan Pablo Ramirez-Hinojosa1, Pablo Maravilla1,*