Abstract
The dense granule protein of Toxoplasma gondii, inhibitor of signal transducer and activator of transcription 1 (IST) is an inhibitor of signal transducer and activator of transcription 1 (STAT1) transcriptional activity that binds to STAT1 and regulates the expression of inflammatory molecules in host cells. A sterile inflammatory liver injury in pathological acute liver failures occurs when excessive innate immune function, such as the massive release of IFN-γ and TNF-α, is activated without infection. In relation to inflammatory liver injury, we hypothesized that Toxoplasma gondii inhibitor of STAT1 transcription (TgIST) can inhibit the inflammatory response induced by activating the STAT1/IRF-1 mechanism in liver inflammation. This study used IFN-γ and TNF-α as inflammatory inducers at the cellular level of murine hepatocytes (Hepa-1c1c7) to determine whether TgIST inhibits the STAT1/IRF-1 axis. In stable cells transfected with TgIST, STAT1 expression decreased with a decrease in interferon regulatory factor (IRF)-1 levels. Furthermore, STAT1 inhibition of TgIST resulted in lower levels of NF-κB and COX2, as well as significantly lower levels of class II transactivator (CIITA), iNOS, and chemokines (CLXCL9/10/11). TgIST also significantly reduced the expression of hepatocyte proapoptotic markers (Caspase3/8/9, P53, and BAX), which are linked to sterile inflammatory liver injury. TgIST also reduced the expression of adhesion (ICAM-1 and VCAM-1) and infiltration markers of programmed death-ligand 1 (PD-L1) induced by hepatocyte and tissue damage. TgIST restored the cell apoptosis induced by IFN-γ/TNF-α stimulation. These results suggest that TgIST can inhibit STAT1-mediated inflammatory and apoptotic responses in hepatocytes stimulated with proinflammatory cytokines.
-
Key words: TgIST, STAT1, acute liver injury, hepatocyte, inflammation, apoptosis
Introduction
Toxoplasma gondii is an obligate intracellular protozoan that can invade and replicate in various warm-blooded animals cells [
1,
2]. After cell invasion by
T. gondii, dense granules discharge secretory proteins that regulate parasitophorous vacuole formation, host cell pathogenicity, and immunity for survival [
3,
4]. Among them, GRA16, GRA24, and an inhibitor of STAT1 transcription (IST) invade the host nucleus and regulate host signaling pathways [
5–
8]. Through these regulations,
T. gondii leaves a distinct transcriptional signature for defense in host cells after infection. As a signal transduction defense,
T. gondii secretes several virulence factors into host cells to inhibit innate and adaptive immune responses [
9]. This immune evasion mechanism enables
T. gondii to survive and proliferate within host cells [
10].
Acute liver failure (ALF) is a complication of drug toxicity, infection, or destructive immune responses [
11]. The liver can be pathologically damaged by the activation of its innate immune function, even in the absence of infection. This is known as “sterile inflammatory liver injury,” and disease examples include drug-induced liver injury (DILI), nonalcoholic steatohepatitis (NASH), and alcoholic steatohepatitis (ASH) [
12]. Increasing evidence suggests that various cytokines play significant roles in the pathogenesis of sterile inflammatory liver injury. The most important proinflammatory cytokines implicated in liver injury development and progression are IFN-γ and TNF-α [
13]. In preclinical and clinical studies of sterile inflammatory liver injury, IFN-γ and TNF-α levels are elevated in the plasma of patients with sterile inflammatory liver injury or experimental animals [
14–
16].
T. gondii-derived inhibitor of STAT1 transcription (TgIST) binds to STAT1 and inhibits its transcriptional function [
17]. STAT1 transmits signals from the engagement of cytokines and growth factors to regulate the transcription of immune-related genes [
17]. STAT1 activation by IFN-γ is a well-known signaling pathway. IFN-γ, a type II interferon, sends signals via STAT1 homodimerization (STAT1/STAT1) to genes with gamma-activated sequences in their promoters [
18]. In response to IFN-γ stimulation, the transcription factor STAT1 induces the transcription of proinflammatory genes that can be regulated by TgIST [
19]. Therefore, STAT1 inhibition by TgIST is a method by which
T. gondii evades host immunity within host cells and can alleviate tissue damage caused by drug toxicity or an unexpectedly excessive inflammatory response.
In this study, we investigated whether TgIST can downregulate proinflammatory gene expression and suppress hepatocyte apoptosis during acute inflammation caused by IFN-γ and TNF-α at the cellular level. This study’s findings suggest that TgIST is a new candidate protein for preventing acute inflammatory liver injury.
Materials and Methods
Ethics statement
Cloning of the TgIST gene in T. gondii and subsequent retrovirus transduction were performed at biosafety level 2 with the approval of the Institutional Biosafety Committee of Seoul National University (IBC SNU; approval number SNUIBC-R180523-1).
Cells
The murine hepatoma cell line (Hepa-1c1c7) and human fibroblast cell line (HFF) were obtained from the Korean Cell Line Bank (Seoul, Korea). The retrovirus packaging cell line (Phoenix) was purchased from Cell Biolabs (San Diego, CA, USA). Hepa-1c1c7, HFF, and Phoenix cells were cultured in high-glucose Dulbecco’s modified Eagle medium (DMEM; WELGENE Inc., Gyeongsan, Korea) containing 10% fetal bovine serum (FBS; WELGENE) and 1% antibiotic–antimycotic (AA) solution (WELGENE) at 37°C in a 5% CO2 incubator. To induce an inflammatory environment in hepatocytes, Hepa-1c1c7 vector cells and Hepa-1c1c7 TgIST cells were seeded in a 6-well plate at 3×105 cells/well and incubated for 24 h. The cells were stimulated with 100 ng/ml of IFN-γ/TNF-α for 24 h before conducting the experiment.
Reagents and chemicals
Murine IFN-γ and TNF-α were supplied by Peprotech (Rocky Hill, NJ, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Kumamoto, Japan).
Toxoplasma gondii
The T. gondii RH strain was obtained from ATCC (Manassas, VA, USA). Tachyzoites of the T. gondii RH strain were serially passaged into HFF cells.
Preparation of a TgIST-expressing Hepa-1c1c7 cell line using a retrovirus system
The TgIST gene sequence (ToxoDB database, Gene ID: TGRH88_0887790) with BamHI (GGATCC) and EcoRI (GAATTC) restriction sites was amplified through PCR using the cDNA of the T. gondii RH strain. The forward primer sequence was 5′-CCGGATCCCGATGCGTTGGTGTGCC-3′, and the reverse primer sequence was 5′-CCGAATTCCTAGACC CGCCGTTCACTAC-3′. PCR was conducted under the following conditions: 95°C for 5 min, followed by 35 cycles of 95°C for 1 min, 60°C for 1 min, and 72°C for 3 min, with a final extension step at 72°C for 10 min. The PCR product (2,691 bp) of TgIST was determined via gene sequencing (Cosmogenetech, Seoul, Korea). To create a TgIST-expressing retrovirus plasmid, the amplified TgIST PCR products and pBABE-HAII plasmid (Addgene, Watertown, MA, USA) were digested with restriction enzymes (BamHI and EcoRI) and ligated with T4 DNA Ligase (Sigma-Aldrich, St. Louis, MO, USA). Phoenix retrovirus packaging cells (Cell Biolabs, San Diego, CA, USA) were transfected with either a plasmid vector (pBABE-HAII) alone or pBABE-HAII-TgIST using a Lipofectamine 3000 transfection kit (Life Technologies, Carlsbad, CA, USA) and Opti-Minimal Essential Medium (Life Technologies). Retrovirus-containing supernatants were centrifuged at 2,000 rpm for 15 min at 4°C and filtered through 0.45-μm syringe filters. To generate a Hepa-1c1c7 cell line stably expressing TgIST, pBABE-HAII-Vector or pBABE-HAII-TgIST retrovirus-containing supernatants were treated with 8 μg/ml of polybrene (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and incubated for 48 h. The retrovirus-infected Hepa-1c1c7 cells were then treated with puromycin (2 μg/ml) (Santa Cruz Biotechnology). TgIST expression in Hepa-1c1c7 cells was measured through PCR using the SureCycler 8800 thermal cycler (Agilent Technologies, Santa Clara, CA, USA), and the PCR products were visualized through 1% agarose gel electrophoresis using the Gel Doc XR+ Gel documentation system (Bio-Rad, Hercules, CA, USA).
Confirmation of stable TgIST expression in the established TgIST-expressing Hepa-1c1c7 cell line
Stable
TgIST expression in Hepa-1c1c7 TgIST cells was confirmed through PCR using
TgIST and/or
pBABE-HAII-Vector primers, including the primer sequences listed in
Supplemenaty Table S1. Total RNA was extracted using the HiGene Total RNA Prep kit (BIOFACT, Daejeon, Korea) and reverse-transcribed into cDNA using the Reverse-Transcription Master Premix kit and oligo d(T)15 primers (ELPIS Biotech, Daejeon, Korea). The cDNA was subjected to PCR to validate
TgIST expression using a SureCycler 8800 thermal cycler (Agilent Technologies, Santa Clara, CA, USA), followed by 1% agarose gel electrophoresis with Stay Safe Nucleic Acid Gel Stain (Real Biotec Corporation, Banqiao City, Taiwan).
Hepa-1c1c7 vector cells and Hepa-1c1c7 TgIST cells were seeded in a 96-well plate at 3×103 cells/well and incubated for 0, 24, 48, 72, and 96 h. The optical density (450 nm, Infinite M200 PRO microplate reader (Tecan)) was measured using the CCK-8 assay kit.
Real-time qPCR
Total RNA was extracted using the HiGene Total RNA Prep Kit (BIOFACT, Daejeon, Korea) and reverse-transcribed to cDNA using the Reverse-Transcription Premix kit (Elpis Biotech, Daejeon, Korea). Real-time qPCR was conducted using the CFX96 Touch Real-Time PCR detection system (Bio-Rad Laboratories) and TOPreal qPCR 2X PreMIX (SYBR Green with low ROX) (Enzynomics, Daejeon, Korea). Amplification reactions were performed at 95°C for 15 min, followed by 40 cycles of 95°C for 20 sec, 59°C for 20 sec, and 72°C for 30 sec. SYBR green fluorescent signals were assessed using the iQTM5 optical system software (Bio-Rad Laboratories). The relative expression of target genes was compared with that of the control group after normalizing to C
t values of
GAPDH. Except for the
P53 primer sequence, the target gene primer sequences were obtained from OriGene Technologies (Rockville, MD, USA). Bassiony et al. [
20] provided the P53 primer sequence.
Supplementary Table S1 shows the target gene primer sequences.
Western blotting was performed using whole-cell lysates from Hepa-1c1c7 stable cells (vector and HA-tagged TgIST). Whole-cell proteins were extracted using M-PERTM mammalian protein extraction reagents (Thermo Fisher Scientific, Waltham, MA, USA). The extracted proteins were quantified using the Pierce BCA protein assay kit (Thermo Fisher Scientific). Proteins were separated using 10% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes at 80 V for 15 min and 150 V for 1 h under ice-cold conditions using a Mini Trans-Blot Electrophoretic Transfer Cell (Bio-Rad). To block unspecific binding of transferred membranes, the membranes were coated with 5% BSA in Tris-buffered saline (TBS-T) (pH 7.6) with 0.1% Tween-20 for 1 h at room temperature (RT). The blocked membranes were incubated with anti-HA tag antibody (Cell Signaling Technology), anti-STAT1 antibody (Santa Cruz Biotechnology), or anti-β-Actin antibody (Santa Cruz Biotechnology) for 16 h at 4°C. The membranes were then washed thrice with TBS-T before incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (m-IgGK BP-HRP; Santa Cruz Biotechnology or Goat anti-Rabbit IgG (H+L)-HRP; Invitrogen) for 1 h at RT. Targets on the membrane were visualized using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). The images were captured using Amersham Imager 600 (GE Healthcare, Chicago, IL, USA), and signal intensities were calculated using ImageJ (Free software provided by the National Institutes of Health).
Immunoprecipitation (IP)
Total proteins were isolated from 2×106 cells of Hepa-1c1c7 stable cells (vector and HA-tagged TgIST) using 400 μl of M-PER mammalian protein extraction reagent (Pierce, Rockford, IL, USA). Next, 100 μl of protein A/G-plus agarose beads (Santa Cruz, Biotechnology) were prereacted with 5 μl of anti-HA tag Ab (Cell Signaling) for 16 h at 4°C. Subsequently, 0.5 mg of total protein extract was added and incubated for 4 h at 4°C. After the supernatants were removed, the anti-HA tag Ab-conjugated protein A/G beads were analyzed through western blotting using anti-STAT1 Ab (Santa Cruz, Biotechnology) and anti-β-Actin Ab (Santa Cruz Biotechnology).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad, La Jolla, CA, USA). The Wilcoxon matched-pairs signed-rank test was used to analyze real-time qPCR data in Hepa-1c1c7 stable cells. A one-way ANOVA followed by a Bonferroni post hoc comparison test was used for the data analysis of real-time qPCR in IFN-γ- or TNF-α-treated Hepa-1c1c7 vector cells. A two-way ANOVA followed by a Bonferroni post hoc comparison test was used to analyze the viability of Hepa-1c1c7 stable cells. Data are expressed as mean±standard deviation (SD). The following significance was assigned: *significant at P<0.05 difference between Hepa-1c1c7 vector cells and Hepa-1c1c7 TgIST cells; †significant at P<0.05 difference between nontreated Hepa-1c1c7 vector cells and IFN-γ-, TNF-α-, and Both-treated Hepa-1c1c7 vector cells.
Results
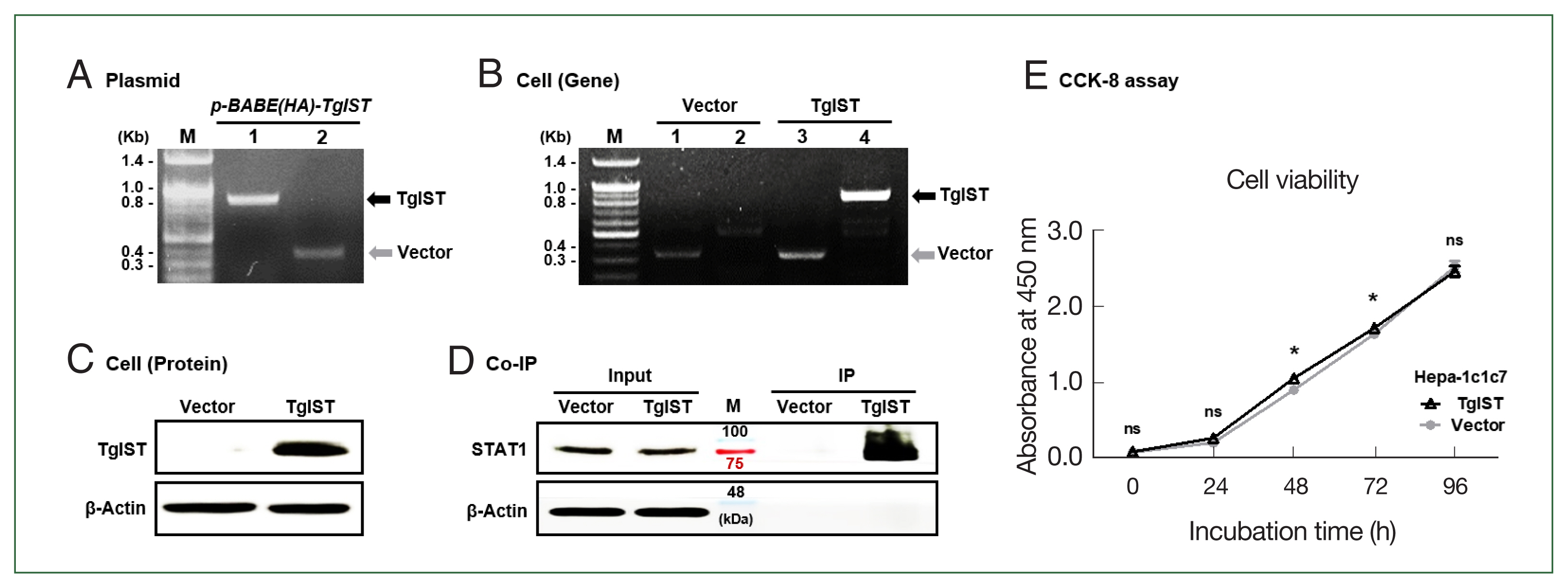
Production of TgIST-expressing Hepa-1c1c7 cells
To create the
TgIST-expressing retroviral vector, the
TgIST (2,673 bp) gene was inserted into the
pBABE-HAII plasmid and confirmed using conventional PCR (
Fig. 1A).
TgIST expression in Hepa-1c1c7 cells was confirmed using PCR and western blotting (
Fig. 1B and C). IP assay revealed a sustained binding interaction between TgIST and STAT1 in Hepa-1c1c7 stable cells (
Fig. 1D). The effects of TgIST on cell proliferation were investigated using the CCK-8 assay. Intracellular TgIST expression had no significant effect on the viability of Hepa-1c1c7 cells (
P<0.05,
Fig. 1E). These results showed that Hepa-1c1c7 cells were continuously expressed TgIST. In the basal state of hepatocytes, TgIST does not affect cell viability (
Fig. 1E).
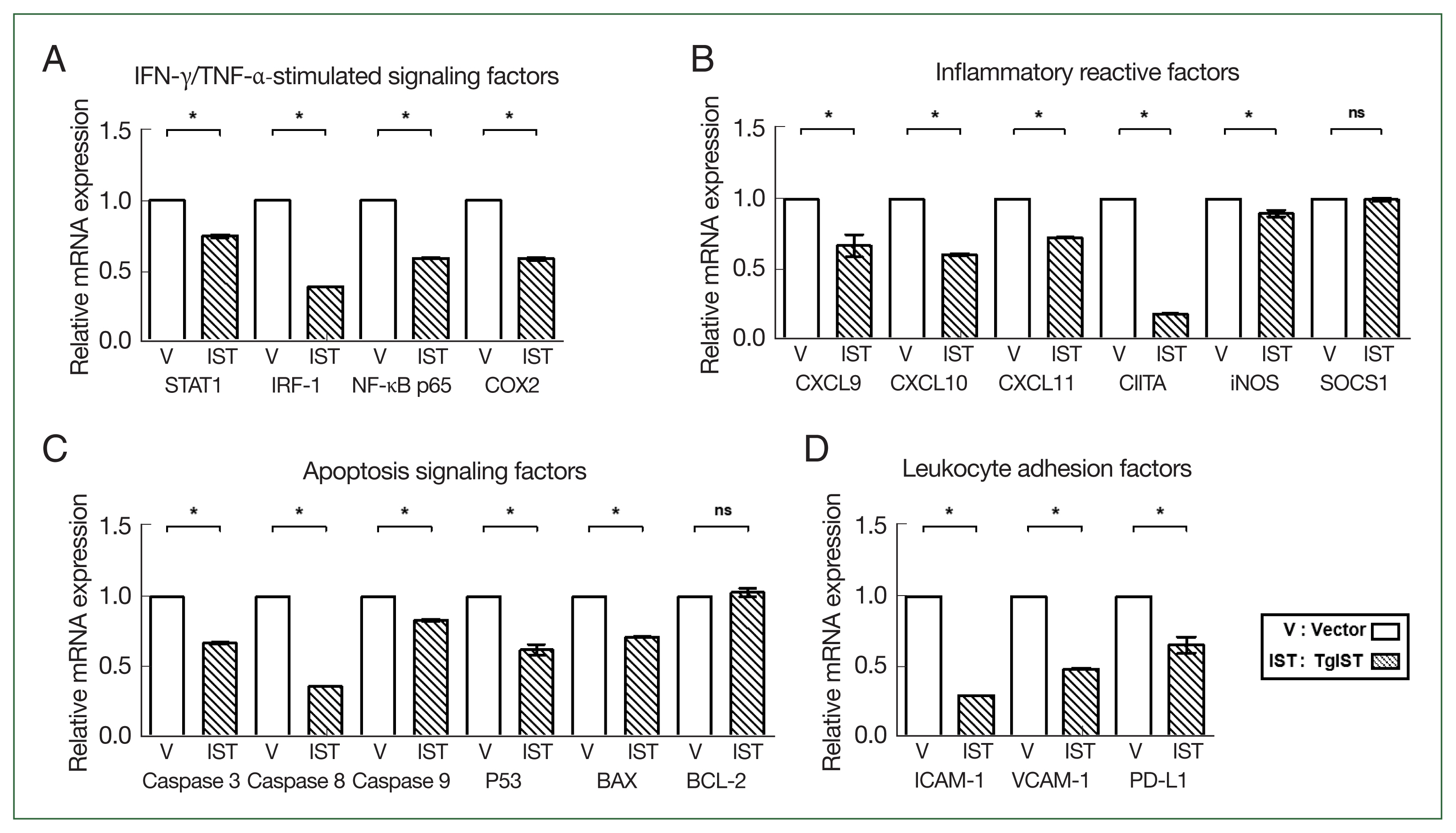
We compared the expression of STAT1-mediated inflammatory and apoptotic genes in Hepa-1c1c7 stable cells. In IFN-γ/TNF-α-stimulated factors, the expression of STAT1, interferon regulatory factor 1 (IRF-1), nuclear factor kappa B (NF-κB), and cyclooxygenase 2 (COX2) was significantly reduced in TgIST cells (
P<0.05,
Fig. 2A). CXC motif chemokine ligands (CXCLs), class II major histocompatibility complex transactivator (CIITA), inducible nitric oxide synthase (iNOS), and suppressor of cytokine signaling 1 (SOCS1) were also studied in response to STAT1 activation. The expression levels of CXCL9/10/11, CTIIA, and iNOS were significantly lower in TgIST cells (
P<0.05,
Fig. 2B). However, SOCS1, which inhibits the biological effects of proinflammatory cytokines by activating the janus kinase (JAK)/STAT1 pathway, differed insignificantly (
P<0.05,
Fig. 2B). After analyzing the expression of caspases, P53, B-cell lymphoma-2 (BCL-2), and BCL-2-associated X (BAX) related to hepatocyte apoptosis occurring in sterile inflammatory liver injury, the proapoptotic molecules (Caspase 3/8/9, P53, and BAX) were significantly increased in TgIST cells (
P<0.05,
Fig. 2C). In contrast, BCL-2, which is crucial in promoting cell survival and inhibiting the action of proapoptotic proteins, was not changed (
P<0.05,
Fig. 2C). Excessive inflammation damages hepatocytes, increasing hepatic adhesion and leukocyte infiltration. The expression of adhesion markers (Intracellular adhesion molecule 1; ICAM-1 and vascular cell adhesion molecule 1; VCAM-1) and infiltration markers (Programmed death-ligand 1; PD-L1) was significantly lower in TgIST cells (
P<0.05,
Fig. 2D). These results show that TgIST suppresses the STAT1-mediated proinflammatory and proapoptotic response by binding to and inhibiting STAT1.
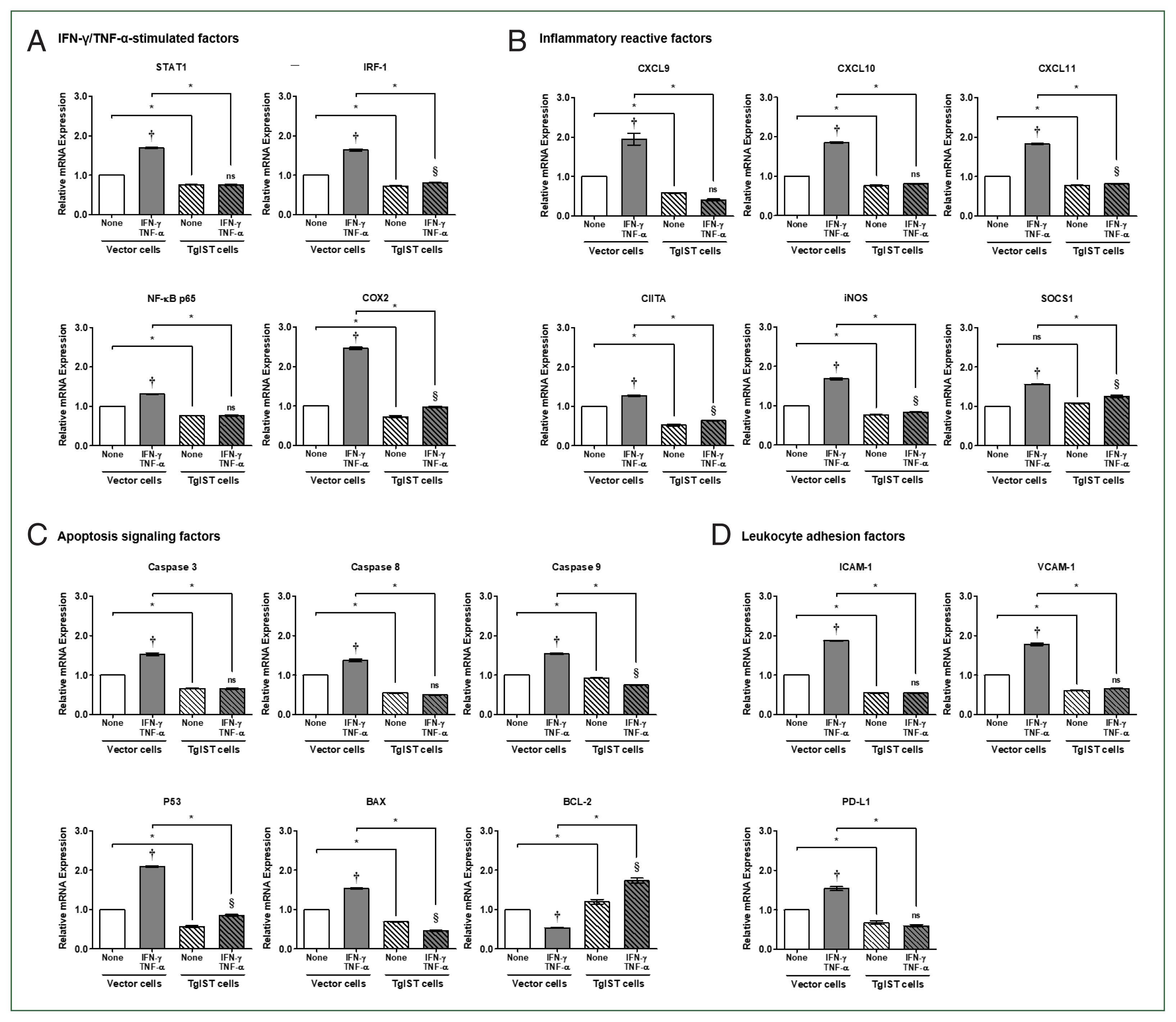
The cellular concentration of IFN-γ/TNF-α treatment was evaluated to provide hepatocytes with an enhanced IFN-γ/TNF-α environment for sterile inflammatory liver injury. First, vector cells were treated with different concentrations of IFN-γ/TNF-α (0, 100, 200, 400, 800, and 1,000 ng/ml) before analyzing the expression of STAT1 and IRF-1. Given the changes in gene expression based on concentration, 100 ng/ml was determined as the final concentration to be employed in the following tests (Data not shown). Hepa-1c1c7 stable cells were treated with a previously determined dose of 100 ng/ml IFN/TNF, and TgIST was tested to determine whether it could continue downregulating STAT1-mediated signaling genes in an IFN/TNF-stimulated environment.
In the case of IFN-γ/TNF-α-stimulated factors, the expression of all genes was considerably elevated in IFN-γ/TNF-α-stimulated vector cells (
P<0.05,
Fig. 3A). In contrast, in TgIST cells, STAT1 and NF-κB did not increase even after IFN-γ/TNF-α stimulation. IRF-1 and COX2 levels increased slightly, but the difference was insignificant (
P<0.05,
Fig. 3A). In STAT1-mediated inflammatory reactive factors, including CXCLs, CIITA, iNOS, and SOCS1, the expression of all genes was also considerably enhanced in IFN-γ/TNF-α-stimulated vector cells (
P<0.05,
Fig. 3B). Except for CXCL9 and CXCL10, TgIST cells increased insignificantly in response to IFN-γ/TNF-α stimulation, although the difference was small (
P<0.05,
Fig. 3B). Although SOCS1 is a negative feedback inhibitor of JAK/STAT cytokine signaling, IFN-γ-mediated STAT1 increases SOCS1 transcription [
21].
Because of assessing the expression of apoptosis signaling factors, proapoptotic genes (
Caspase 3/8/9, P53, and
BAX) were considerably elevated in vector cells (
P<0.05,
Fig. 3C). However, these genes were not altered (
Caspase 3/8), decreased (
Caspase 9), or modestly elevated (
P53) in TgIST cells (
P<0.05,
Fig. 3C). In the case of BCL-2, an antiapoptotic gene was dramatically lowered in IFN-γ/TNF-α-treated vector cells but significantly increased in IFN-γ/TNF-α-stimulated TgIST cells (
P<0.05,
Fig. 3C). Meanwhile, the expression of leukocyte adhesion-related genes (
ICAM-1 and
VCAM-1) and infiltration-related genes (
PD-L1) increased in vector cells but not in TgIST cells (
P<0.05,
Fig. 3D).
These findings indicate that when hepatocytes are stimulated by IFN-γ/TNF-α, as occurs in sterile inflammatory liver injury, TgIST can continuously suppress the expression of proinflammatory, proapoptotic, and hepatic adhesion/infiltration genes via activated STAT1.
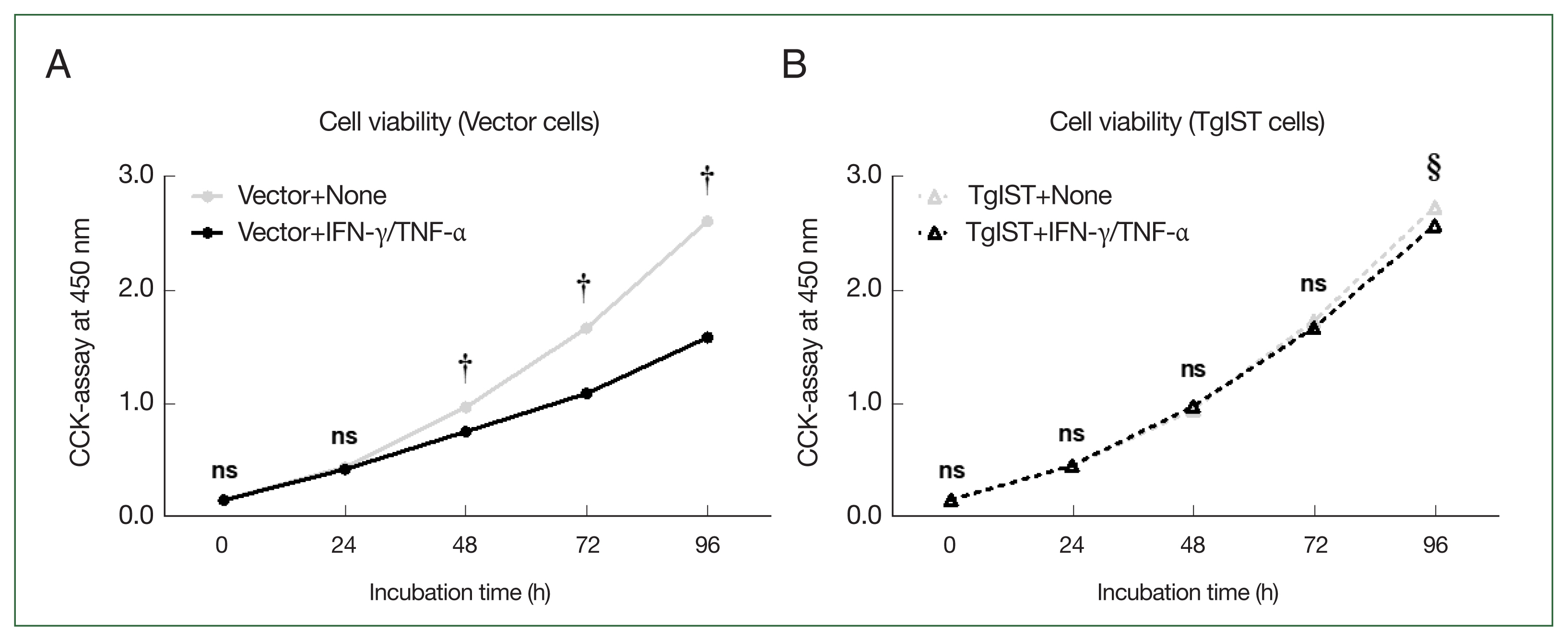
Effect of TgIST on the viability of hepatocytes stimulated with IFN-γ/TNF-α
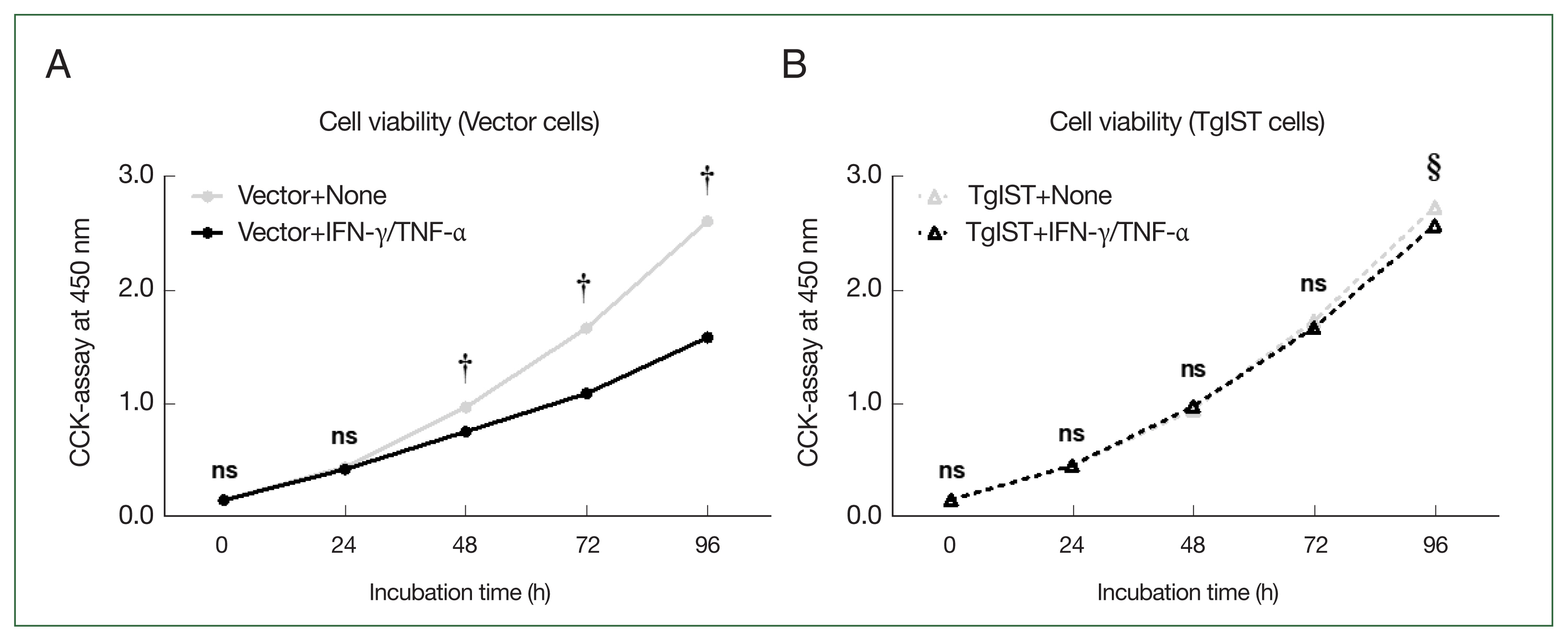
We analyzed whether inhibiting STAT1-mediated signaling mechanisms using TgIST could improve the attenuation of hepatocyte proliferation stimulated by IFN-γ/TNF-α. The CCK-8 assay was used to assess the viability of Hepa-1c1c7 stable cells over time after stimulation with IFN-γ/TNF-α (
Fig. 4). In vector cells, cell proliferation was significantly lower than that in the nonstimulated group (Vector+none) starting at 48 h after stimulation with IFN-γ/TNF-α (
P<0.05,
Fig. 4A). In contrast, in TgIST cells, cell proliferation differed insignificantly from that in the nonstimulated group (TgIST+none) at any time point despite stimulation with IFN-γ/TNF-α (
P<0.05,
Fig. 4B). Therefore, TgIST improves the attenuated hepatocyte proliferation induced by IFN-γ/TNF-α stimulation and sustains cell growth.
Discussion
In this study, inflammatory and apoptotic gene expression changes after IFN-γ/TNF-α stimulation in TgIST-stable hepatocytes were investigated. Therefore, we produced TgIST gene-expressing hepatocytes using a retrovirus transfection system. Our results showed that the expression of inflammatory and apoptotic genes was suppressed in TgIST-stable cells stimulated with IFN-γ/TNF-α. In addition, TgIST-stable cells were not induced by apoptosis, unlike IFN-γ/TNF-α stimulation, which can induce apoptosis in hepatocytes. We found that TgIST reduces STAT1 signaling. In this study, we found that TgIST inhibited STAT-1-mediated inflammation and cell apoptosis in IFN-γ/TNF-α-stimulated hepatocytes and suggested TgIST as a novel candidate protein for preventing inflammation causing acute inflammatory liver injury.
Many studies have shown that
T. gondii infection causes a robust IFN-γ-stimulated immune response that is important for resolving acute infection and controlling chronic infection, and recent studies have confirmed that TgIST is required to inhibit STAT1-mediated transcription induced by IFN-γ [
17]. Therefore, we hypothesized that TgIST inhibits the mechanism of inflammatory liver injury by inhibiting
STAT1 and
IRF-1 gene expression. A sterile inflammatory liver injury is a condition that causes excessive inflammation and cell damage in the liver but does not result from infection [
12]. As an example, DILI is commonly caused by drugs such as acetaminophen or its metabolites and manifests as liver damage [
22]. In the United States (U.S.), DILI accounts for >50% of patients with ALF; currently, the only treatments clinically approved by the U.S. Food and Drug Administration are hepatoprotective antidotes such as N-acetylcysteine (NAC) and L-carnitine or liver transplantation. However, it is impossible to fundamentally treat the increased inflammatory response and liver cell damage [
23,
24]. DILI is caused by various immune cells, including Kupffer cells, dendritic cells, and natural killer cells, which are associated with the innate immune response [
25]. Excessively activated immune cells primarily release IFN-γ and TNF-α, which have a synergistic effect on the production of various proinflammatory cytokines and chemokines via the STAT1/IRF-1 axis and directly contribute to hepatocellular damage [
13,
26,
27]. Therefore, treatment strategies targeting the STAT1/IRF-1 axis are becoming increasingly important in sterile inflammatory liver injury, for which no targeted treatment mechanism is available [
28].
Therefore, we confirmed that TgIST can downregulate the inflammatory response and apoptosis that occur in sterile inflammatory liver injury at the cellular level via its action mechanism on the IFN-γ/TNF-α induced STAT1/IRF-1 signaling pathway. The mechanism by which STAT1-mediated inflammatory factor expression changes after transfecting
TgIST into Hepa-1c1c7 cells was investigated. In this study, we repeatedly confirmed the protein–protein interaction of TgIST and STAT1 using IP. Furthermore, despite stimulation with IFN-γ/TNF-α, TgIST cells showed lower expression of IFN-γ/TNF-α-stimulated factors (STAT1, IRF-1, NF-κB, and COX2) compared with vector cells. IRF-1 and NF-κB are transcription factors that increase the expression of inflammatory cytokines and apoptosis-related molecules [
29]. Furthermore, COX2 is a proinflammatory stimulus that is expressed via the transcriptional effects of STAT1 and NF-κB [
30]. CXCL9, CXCL10, and CXCL11 are proinflammatory chemokines that exacerbate DILI-induced hepatocyte damage by promoting hepatocyte apoptosis via binding to CXC receptor 3 (CXCR3) [
31]. The expression of proinflammatory chemokines (CXCL9/10/11) and IFN-γ-inducible transcriptional coactivator (CIITA), which was increased in vector cells after IFN-γ/TNF-α stimulation, was consistently suppressed in TgIST cells. Similarly, unrestrained IFN-γ and TNF-α signaling directly induces inflammatory gene expression and causes cell death, inflammatory immune responses, and disease development [
32]. The expression of proapoptotic markers (Caspase3/8/9, P53, and BAX) increased in vector cells when stimulated with IFN-γ/TNF-α, but decreased in TgIST cells. In contrast, the expression of the antiapoptotic marker BCL-2 decreased in vector cells when stimulated with IFN-γ/TNF-α, but increased in TgIST cells.
Drug-induced hepatotoxicity causes rapid recruitment of leukocytes to the inflammation site and activates effective apoptosis mechanisms. However, exorbitant leukocytes can cause acute tissue damage, including liver damage, under various pathophysiological conditions because of their potential cytotoxicity [
33]. Therefore, the expression of adhesion molecules (ICAM-1 and VCAM-1) and infiltration molecules (PD-L1) can be used to detect excessive leukocyte infiltration [
34]. TgIST inhibited ICAM-1, VCAM-1, and PD-L1, which were increased by IFN-γ/TNF-α stimulation.
In this study, we confirmed that TgIST downregulates the expression of proinflammatory and proapoptotic molecules, which are hallmarks of sterile inflammatory liver injury, via the STAT1/IRF-1 axis when stimulated by IFN-γ/TNF-α. Consequently, TgIST prevented cell death and promoted continuous cell proliferation by suppressing decreased cell viability caused by stimulation with IFN-γ/TNF-α. Therefore, these TgIST properties can be used to create targeted therapeutic agents that regulate STAT1/IRF-1 in sterile inflammatory liver injury. For the translational availability of the TgIST anti-inflammation agent, a cell-penetrating peptide (CPP) as a therapeutic agent would be a promising strategy because of its induction of intracellular penetration [
35]. Clinical trials using CPPs have been conducted to test the efficacy of CPP-based treatments [
35]. Although more research is required to determine the efficacy of TgIST in various types of liver injury, this study’s findings highlight TgIST as a promising inflammation inhibitor that can exert anti-inflammatory effects via STAT1/IRF-1 inhibition.
Notes
-
The authors declare no conflicts of interest related to this work.
-
Conceptualization: Seo SH, Shin EH
Data curation: Seo SH, Lee JE, Ham DW, Shin EH
Funding acquisition: Shin EH
Investigation: Seo SH, Lee JE
Methodology: Seo SH, Lee JE, Ham DW
Project administration: Shin EH
Supervision: Shin EH
Validation: Seo SH, Shin EH
Visualization: Seo SH
Writing – original draft: Seo SH, Shin EH
Writing – review & editing: Shin EH
Supplementary Information
Acknowledgments
This work was supported by the National Research Foundation of Korea (No. NRF-2022R1A2C1009234) and also by grant no. 02-2022-0009 from the Seoul National University Bundang Hospital (SNUBH) Research Fund.
Fig. 1Production of TgIST-expressing Hepa-1c1c7 cells. (A) PCR results for the TgIST gene (Lane 1, fragment) or vector gene (Lane 2, fragment) in the pBABE-HAII-TgIST plasmid. (B) PCR results for the vector gene and TgIST gene in Hepa-1c1c7 vector cells (Lane 1, 2) or Hepa-1c1c7 TgIST cells (Lane 3, 4). (C) HA-tagged TgIST protein expression in TgIST cells. (D) Immunoprecipitation (IP) confirms the interaction between TgIST and STAT1. Input: Immunostaining of TgIST in total protein before IP analysis. IP: Western blotting stained with anti-STAT1 ab for the protein fraction (TgIST) extracted using anti-HA tag ab from Hepa-1c1c7 stable cells. (E) Time-dependent viability of Hepa-1c1c7 stable cells. M, marker; ns, not significant; *P<0.05 between vector and TgIST cells.
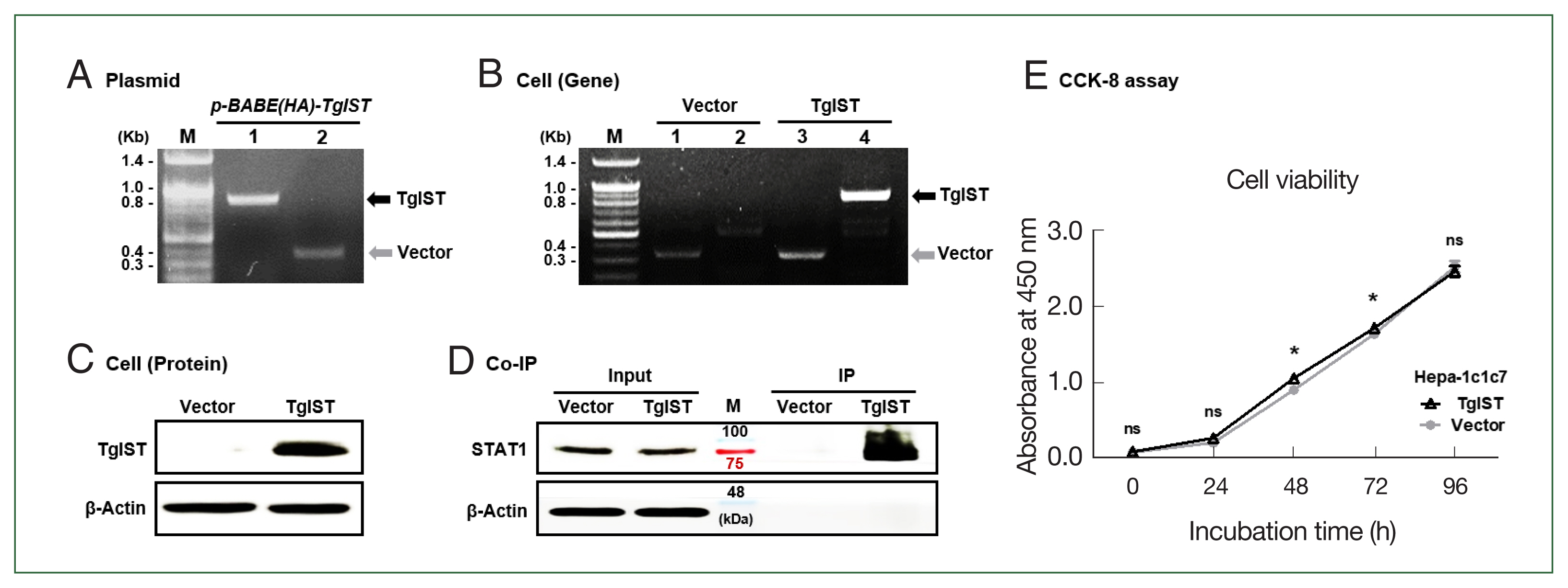
Fig. 2Effect of TgIST on STAT1-mediated inflammatory or apoptotic genes in the basal state of hepatocytes. (A) mRNA expression of IFN-γ/TNF-α-stimulated factors. (B–D) mRNA expression of STAT1-mediated inflammatory reactive factors (B), apoptosis signaling factors (C), and hepatic adhesion factors (D) in Hepa-1c1c7-stable cells. ns, not significant. *P<0.05 between vector and TgIST cells.
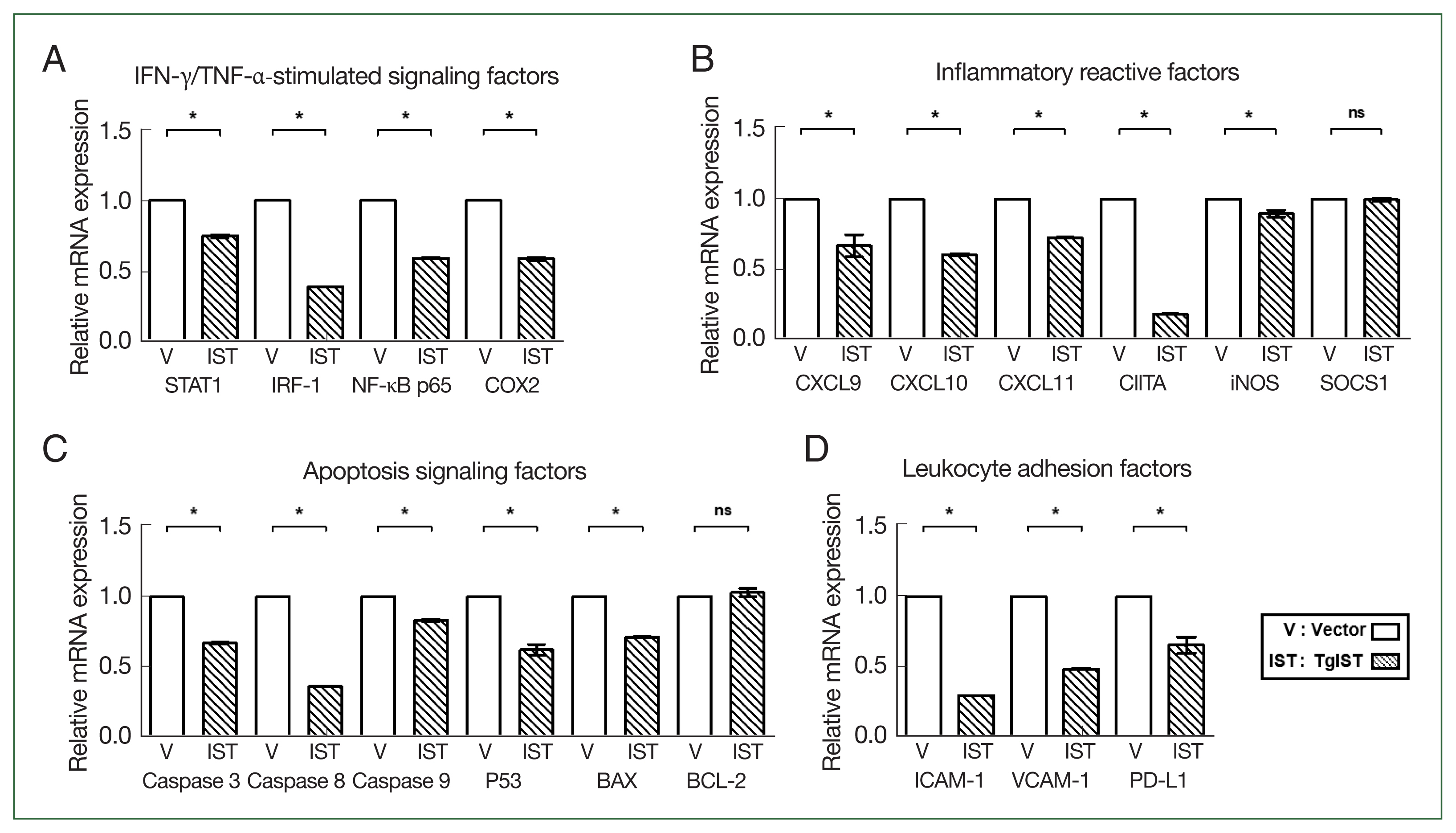
Fig. 3Effect of TgIST on STAT1-mediated inflammatory and apoptotic genes in IFN/TNF-stimulated hepatocytes. (A) mRNA expression of IFN-γ/TNF-α-stimulated factors in IFN-γ/TNF-α-stimulated stable cells. (B–D) mRNA expression of STAT1-mediated inflammatory reactive factors (B), apoptosis signaling factors (C), and hepatic adhesion and infiltration factors (D) in IFN-γ/TNF-α-stimulated stable cells. None, nonstimulated group ns, not significant. †P<0.05 difference between nontreated vector and IFN-γ/TNF-α-stimulated vector, §P<0.05 difference between nontreated TgIST and IFN-γ/TNF-α-stimulated TgIST, *P<0.05 between vector and TgIST cells.
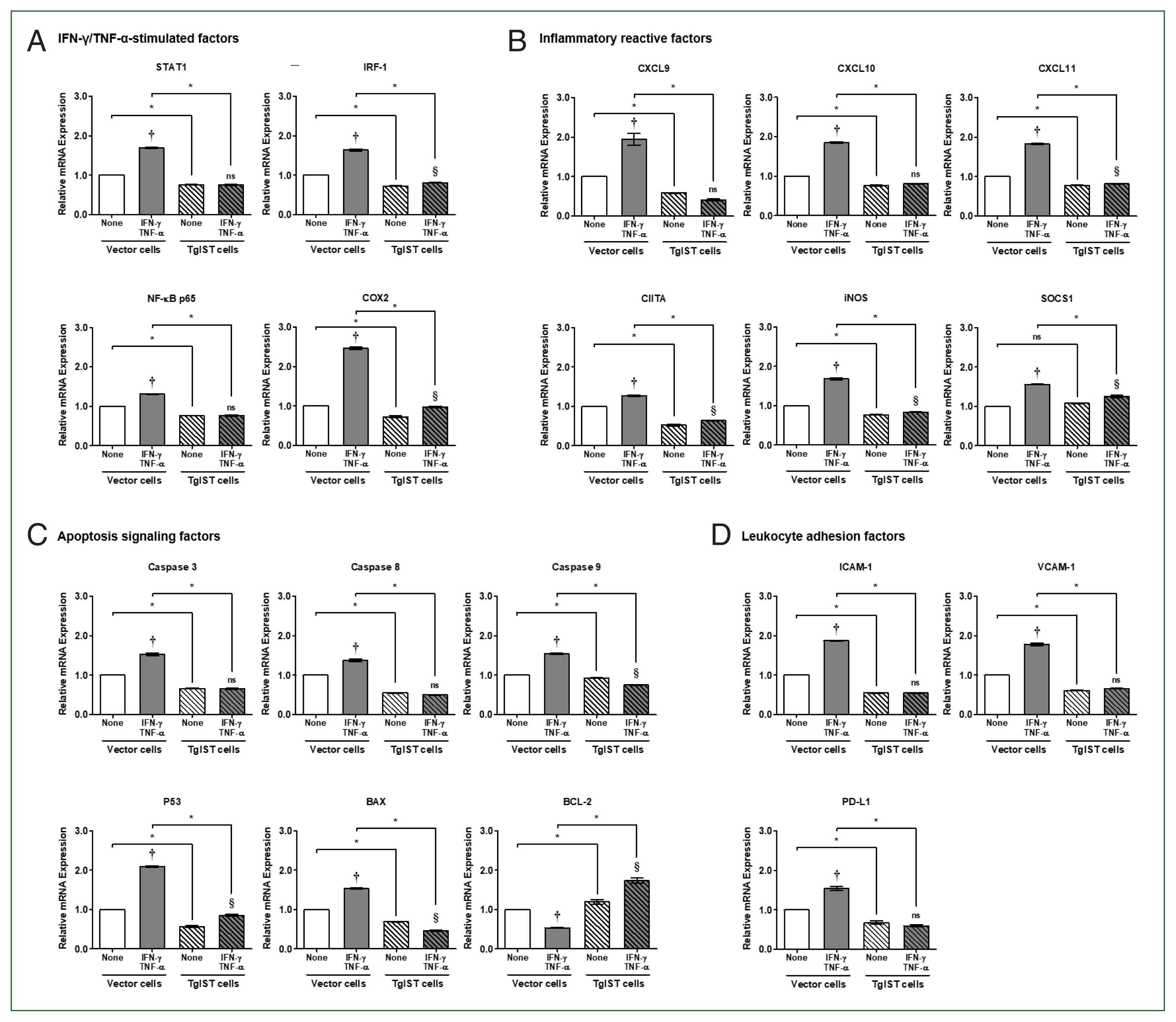
Fig. 4Effect of TgIST on cell viability in hepatocytes stimulated with IFN/TNF. (A) Time-dependent analysis of cell viability after Hepa-1c1c7 vector cells were incubated with IFN-γ/TNF-α (100 ng/ml) for 24, 48, 72, or 96 h. (B) Time-dependent analysis of cell viability after Hepa-1c1c7 TgIST cells were incubated with IFN-γ/TNF-α (100 ng/ml) for 24, 48, 72, or 96 h. None, nonstimulated group; ns, not significant. †P<0.05 difference between nonstimulated vector and IFN-γ/TNF-α-stimulated vector, §P<0.05 difference between nonstimulated TgIST and IFN-γ/TNF-α-stimulated TgIST, *P<0.05 between vector and TgIST cells.
References
- 1. Lee JH, Yuk JM, Cha GH, Lee YH. Expression of cytokines and co-stimulatory molecules in the Toxoplasma gondii-infected dendritic cells of C57BL/6 and BALB/c mice. Parasites Hosts Dis 2023;61(2):138. https://doi.org/10.3347/PHD.22150
- 2. Kim GH, Kwak JH, Park YH. Elevated interleukin-10 levels in patients with ocular toxoplasmosis. Parasites Hosts Dis 2023;61(3):310. https://doi.org/10.3347/PHD.22172
- 3. Seo SH, Shin JH, Ham DW, Shin EH. PTEN/AKT signaling pathway related to hTERT downregulation and telomere shortening induced in Toxoplasma GRA16-expressing colorectal cancer cells. Biomed Pharmacother 2022;153:113366. https://doi.org/10.1016/j.biopha.2022.113366
- 4. Zhang Y, Lai BS, Juhas M, Zhang Y. Toxoplasma gondii secretory proteins and their role in invasion and pathogenesis. Microbiol Res 2019;227:126293. https://doi.org/10.1016/j.micres.2019.06.003.5
- 5. Kim SG, Seo SH, Shin JH, Yang JP, Lee SH, et al. Increase in the nuclear localization of PTEN by the Toxoplasma GRA16 protein and subsequent induction of p53-dependent apoptosis and anticancer effect. J Cell Mol Med 2019;23(5):3234-3245. https://doi.org/10.1111/jcmm.14207
- 6. Seo SH, Kim SG, Shin JH, Ham DW, Shin EH. Toxoplasma gra16 inhibits nf-κb activation through pp2a-b55 upregulation in non-small-cell lung carcinoma cells. Int J Mol Sci 2020;21(18):6642. https://doi.org/10.3390/ijms21186642
- 7. Krishnamurthy S, Saeij JP. Toxoplasma does not secrete the GRA16 and GRA24 effectors beyond the parasitophorous vacuole membrane of tissue cysts. Front Cell Infect Microbiol 2018;8:366. https://doi.org/10.3389/fcimb.2018.00366
- 8. Olias P, Etheridge RD, Zhang Y, Holtzman MJ, Sibley LD. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe 2016;20(1):72-82. https://doi.org/10.1016/j.chom.2016.06.006
- 9. Sasai M, Yamamoto M. Innate, adaptive, and cell-autonomous immunity against Toxoplasma gondii infection. Exp Mol Med 2019;51(12):1-10. https://doi.org/10.1038/s12276-019-0353-9
- 10. Seizova S, Ruparel U, Garnham AL, Bader SM, Uboldi AD, et al. Transcriptional modification of host cells harboring Toxoplasma gondii bradyzoites prevents IFN gamma-mediated cell death. Cell Host Microbe 2022;30(2):232-247. https://doi.org/10.1016/j.chom.2021.11.012
- 11. Xu Y, Wang J, Ren H, Dai H, Zhou Y, et al. Human endoderm stem cells reverse inflammation-related acute liver failure through cystatin SN-mediated inhibition of interferon signaling. Cell Res 2023;33(2):147-164. https://doi.org/10.1038/s41422-022-00760-5
- 12. Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology 2012;143(5):1158-1172. https://doi.org/10.1053/j.gastro.2012.09.008
- 13. Wesemann DR, Benveniste EN. STAT-1α and IFN-γ as modulators of TNF-α signaling in macrophages: regulation and functional implications of the TNF receptor 1: STAT-1α complex. J Immunol 2003;171(10):5313-5319. https://doi.org/10.4049/jimmunol.171.10.5313
- 14. Sutti S, Bruzzì S, Albano E. The role of immune mechanisms in alcoholic and nonalcoholic steatohepatitis: a 2015 update. Expert Rev Gastroenterol Hepatol 2016;10(2):243-253. https://doi.org/10.1586/17474124.2016.1111758
- 15. Woolbright BL, Nguyen NT, McGill MR, Sharpe MR, Curry SC, et al. Generation of pro-and anti-inflammatory mediators after acetaminophen overdose in surviving and non-surviving patients. Toxicol Lett 2022;367:59-66. https://doi.org/10.1016/j.toxlet.2022.07.813
- 16. Tiegs G, Horst AK. TNF in the liver: targeting a central player in inflammation. Semin Immunopathol 2022;44(4):445-459. https://doi.org/10.1007/s00281-022-00910-2
- 17. Olias P, Etheridge RD, Zhang Y, Holtzman MJ, Sibley LD. Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe 2016;20(1):72-82. https://doi.org/10.1016/j.chom.2016.06.006
- 18. Huang Z, Liu H, Nix J, Xu R, Knoverek CR, et al. The intrinsically disordered protein TgIST from Toxoplasma gondii inhibits STAT1 signaling by blocking cofactor recruitment. Nat Commun 2022;13(1):4047. https://doi.org/10.1038/s41467-022-31720-7
- 19. Krause CD, He W, Kotenko S, Pestka S. Modulation of the activation of Stat1 by the interferon-γ receptor complex. Cell Res 2006;16(1):113-123. https://doi.org/10.1038/sj.cr.7310015
- 20. Bassiony H, Sabet S, Salah El-Din TA, Mohamed MM, El-Ghor AA. Magnetite nanoparticles inhibit tumor growth and upregulate the expression of P53/P16 in Ehrlich solid carcinoma bearing mice. PLoS One 2014;9(11):e111960. https://doi.org/10.1371/journal.pone.0111960
- 21. Davey GM, Heath WR, Starr R. SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation. Tissue Antigens 2006;67(1):1-9. https://doi.org/10.1111/j.1399-0039.2005.00532.x
- 22. Liu W, Zeng X, Liu Y, Liu J, Li C, et al. The immunological mechanisms and immune-based biomarkers of drug-induced liver injury. Front Pharmacol 2021;12:723940. https://doi.org/10.3389/fphar.2021.723940
- 23. Hosack T, Damry D, Biswas S. Drug-induced liver injury: a comprehensive review. Therap Adv Gastroenterol 2023;16:17562848231163410. https://doi.org/10.1177/17562848231163410
- 24. Katarey D, Verma S. Drug-induced liver injury. Clin Med (Lond) 2016;16(suppl):104-109. https://doi.org/10.7861/clinmedicine.16-6s-s104
- 25. Tasnim F, Huang X, Lee CZW, Ginhoux F, Yu H. Recent advances in models of immune-mediated drug-induced liver injury. Front Toxicol 2021;3:605392. https://doi.org/10.3389/ftox.2021.605392
- 26. Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 2021;184(1):149-168. https://doi.org/10.1016/j.cell.2020.11.025
- 27. Bradham CA, Plümpe J, Manns MP, Brenner DA, Trautwein C. Mechanisms of hepatic toxicity. I. TNF-induced liver injury. Am J Physiol 1998;275(3):G387-G392. https://doi.org/10.1152/ajpgi.1998.275.3.G387
- 28. Tomita K, Kabashima A, Freeman BL, Bronk SF, Hirsova P, et al. Mixed lineage kinase 3 mediates the induction of CXCL10 by a STAT1-dependent mechanism during hepatocyte lipotoxicity. J Cell Biochem 2017;118(10):3249-3259. https://doi.org/10.1002/jcb.25973
- 29. Iwanaszko M, Kimmel M. NF-κB and IRF pathways: cross-regulation on target genes promoter level. BMC Genomics 2015;16(1):1-8. https://doi.org/10.1186/s12864-015-1511-7
- 30. Shanmugam N, Kim YS, Lanting L, Natarajan R. Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J Biol Chem 2003;278(37):34834-34844. https://doi.org/10.1074/jbc.M302828200
- 31. Song X, Shen Y, Lao Y, Tao Z, Zeng J. CXCL9 regulates acetaminophen-induced liver injury via CXCR3. Exp Ther Med 2019;18(6):4845-4851. https://doi.org/10.3892/etm.2019.8122
- 32. van Loo G, Bertrand MJ. Death by TNF: a road to inflammation. Nat Rev Immunol 2023;23(5):289-303. https://doi.org/10.1038/s41577-022-00792-3
- 33. Jaeschke H, Ramachandran A. Mechanisms and pathophysiological significance of sterile inflammation during acetaminophen hepatotoxicity. Food Chem Toxicol 2020;138:111240. https://doi.org/10.1016/j.fct.2020.111240
- 34. Gerussi A, Natalini A, Antonangeli F, Mancuso C, Agostinetto E, et al. Immune-mediated drug-induced liver injury: immunogenetics and experimental models. Int J Mol Sci 2021;22(9):4557. https://doi.org/10.3390/ijms22094557
- 35. Bottens RA, Yamada T. Cell-penetrating peptides (CPPs) as therapeutic and diagnostic agents for cancer. Cancers (Basel) 2022;14(22):5546. https://doi.org/10.3390/cancers14225546