Abstract
Plasmodium falciparum apical membrane antigen-1 (PfAMA-1) is a major candidate for the blood-stage malaria vaccine. Genetic polymorphisms of global pfama-1suggest that the genetic diversity of the gene can disturb effective vaccine development targeting this antigen. This study was conducted to explore the genetic diversity and gene structure of pfama-1 among P. falciparum isolates collected in the Khyber Pakhtunkhwa (KP) province of Pakistan. A total of 19 full-length pfama-1 sequences were obtained from KP-Pakistan P. falciparum isolates, and genetic polymorphism and natural selection were investigated. KP-Pakistan pfama-1 exhibited genetic diversity, wherein 58 amino acid changes were identified, most of which were located in ectodomains, and domains I, II, and III. The amino acid changes commonly found in the ectodomain of global pfama-1 were also detected in KP-Pakistan pfama-1. Interestingly, 13 novel amino acid changes not reported in the global population were identified in KP-Pakistan pfama-1. KP-Pakistan pfama-1 shared similar levels of genetic diversity with global pfama-1. Evidence of natural selection and recombination events were also detected in KP-Pakistan pfama-1.
-
Key words: Plasmodium falciparum, apical membrane antigen-1, genetic diversity, Pakistan
Introduction
Malaria caused by
Plasmodium species is a major infectious disease in humans, causing a significant global public health burden. Irrespective of the massive control efforts to eliminate malaria over the past years, malaria is still prevalent in several endemic areas. The World Health Organization reported 247 million malaria cases and 619,000 deaths in 2021 [
1]. Malaria control and elimination efforts have been challenged due to the spread of antimalarial drug-resistant parasites and insecticide-resistant
Anopheles mosquitoes. The lack of an effective vaccine creates a major obstacle in malaria control, indicating the indispensable need to develop an effective vaccine. Several plasmodial proteins such as circumsporozoite protein (CSP), Duffy binding protein, merozoite surface proteins, apical membrane antigen-1 (AMA-1), and thrombospondin-related anonymous protein have been considered promising vaccine candidates because of their antigenic properties and expression in either preerythrocytic or erythrocytic stages of malaria parasites [
2,
3]. However, the genetic polymorphisms in these antigens among clinical isolates are significant hurdles in the development of effective malaria vaccines [
4,
5].
AMA-1 is a type I integral membrane protein commonly expressed on the surfaces of merozoites and sporozoites and plays important roles in parasite invasion into host cells [
6,
7]. It comprises a signal sequence, a cysteine-rich ectodomain, a conserved cytoplasmic region, and a transmembrane region [
8]. The ectodomain of AMA-1 is further subdivided into 3 domains, viz., domains I (DI), II (DII), and III (DIII). The ectodomain is highly immunogenic and induces natural immune responses in individuals infected with
P. falciparum [
9,
10]. Antibodies against AMA-1 prevent the invasion of erythrocytes by malaria parasites and establish a protective immune response [
11,
12]. This information suggests that the antigen is a feasible vaccine candidate. Similar to that in other major surface antigens of malaria parasites, substantial levels of genetic polymorphisms of
ama-1 have also been recognized in wild parasite populations; however, AMA-1 has been considered less variable than other potential vaccine candidate antigens such as CSP and merozoite surface proteins, supporting the notion that it is a promising blood-stage vaccine candidate [
10,
13,
14]. Nevertheless, genetic diversities observed in global
ama-1 have emphasized the importance of continuous monitoring of genetic variations of
ama-1 among global malaria parasites for designing an effective vaccine targeting this antigen [
15].
Pakistan is a malaria-endemic country, with reports of millions of cases per year [
16].
P. falciparum and
P. vivax are the prevalent species, accounting for 32% and 67% of malaria cases, respectively. Khyber Pakhtunkhwa (KP) and Balochistan provinces have been the most critical malaria hot spots in the country [
16]. A study on the genetic analysis of the hypervariable DI of
P. falciparum AMA-1 (
pfama-1) in Pakistan
P. falciparum isolated in Hazara division, KP, Pakistan, had been performed [
17]. However, the sequences did not cover full-length gene sequences, rendering only limited information on the genetic nature of Pakistan
pfama-1. This study was conducted to analyze the genetic nature of full-length
pfama-1 among
P. falciparum isolates collected from the KP province of Pakistan.
Materials and Methods
Ethics statement
The study protocol was approved by the Ethical Review Committee of Abdul Wali Khan University Mardan under the letter of AWKUM/ERC/578. Consent was obtained from all participants before conducting the study.
Parasite samples and DNA purification
Blood samples were collected from 19 patients infected with
P. falciparum, which was confirmed by microscopy and rapid diagnostic tests in different hospitals and private sector laboratories in KP, Pakistan (
Supplementary Fig. S1). During the 2 malaria seasons from March to May and August to November 2019, the area had an average annual rainfall of 384 mm. The mean temperature in the region was 20°C–40°C. The blood samples collected before treatment were spotted on filters, air-dried, and stored in individually sealed plastic bags at ambient temperature until use. Genomic DNA was extracted from the spotted blood samples using a QIAamp blood mini kit (Qiagen, Redwood City, CA, USA) according to the manufacturer’s instructions and stored at −20°C.
The full-length
pfama-1 was amplified by PCR using specific primer sets and amplification conditions described previously [
18,
19]. The PCR products were analyzed on a 1.5% agarose gel, purified, and cloned into the T&A vector (Real Biotech, Banqiao City, Taiwan). The ligation mixture was transformed into
Escherichia coli DH5α competent cells, and positive clones were selected by colony PCR. The nucleotide sequence of the cloned insert was analyzed by automatic DNA sequencing with M13 forward and M13 reverse primers (Genotech, Daejeon, Korea). Sequencing was also conducted using 2 additional internal primers (5′-CAGGGAAATGTCCAGTATTTGGTA-3′ and 5′-TTCCATCGACCCATAATCCG-3′) to obtain clear sequences for the central part of
pfama-1 [
18]. To ensure accuracy, sequencing of at least 2 different clones from each isolate was performed. Raw data were filtered for quality assessment using the DNASTAR Lasergene software (DNASTAR, Madison, WI, USA). The 19 KP-Pakistan
pfama-1 nucleotide sequences were deposited in GenBank under accession numbers OM628702–OM628720.
The DNA sequence generated in this study was analyzed in comparison with a reference gene of
pfama-1 from the
P. falciparum 3D7 strain (GenBank Accession No.: U65407). The following global
pfama-1 sequences deposited in GenBank were also included for analysis: Thailand (AB715735–AB715814), Myanmar (KU893276–KU893333), Philippines (AB715815–AB715869), Vietnam (MW938322–MW938452), Vanuatu (AB716010–AB716094), Solomon Islands (SI; AB715960–AB716009), Papua New Guinea (PNG; AB715870–AB715959), Ghana (AB715698–AB715734), and Tanzania (AB715636–AB715697) (
Supplementary Table S1). Comparative sequence analyses were conducted to identify polymorphic loci using the MEGA6 program [
20].
The DnaSP v6.12 software package [
21] was used to estimate parsimony informative sites, total number of mutations, pairwise nucleotide diversity (π), haplotype diversity, segregating sites, haplotypes, recombination between adjacent nucleotides per generation, and the minimum number of recombination events (Rm). Linkage disequilibrium was estimated between the various polymorphic sites based on the recombination events (
R2) index using the DnaSP v6.12 software package [
21]. Tajima’s D, Fu and Li’s D, and F indices were calculated by a sliding window method using the DnaSP v6.12 software package [
21]. Population genetics, including pairwise fixation index (
FST) and haplotype frequencies, were evaluated using the analysis of molecular variance. The significance of the analysis of molecular variance was estimated by 1,000 per mutation, and the nucleotide diversity based on Nei’s net distance was computed using Arlequin v3.5 [
22]. The haplotype network plot was generated using the PopArt software [
23].
The global
pfama-1 sequences were aligned and filtered using the GUIDANCE server with the confidence score threshold (i.e., best score ~1) [
24]. This low-quality alignment filtration is essential for the accuracy of the natural selection analysis [
25]. The good-quality, reliable alignment was subjected to the Datamonkey server of the HYPHY package for the identification of selected loci with a default
P value [
26–
28]. Individual sites underlying positive selection were inferred using the following 3 algorithms: fixed effects likelihood, internal branches fixed effects likelihood, and mixed effects model of evolution [
28].
Results
Genetic polymorphic features of KP-Pakistan pfama-1
The 19 full-length
pfama-1 sequences were successfully amplified from the 19 KP-Pakistan
P. falciparum isolates. The gene length was 1,869 bp, and no size polymorphism was identified in the sequences. Comparative analysis of the 19 KP-Pakistan
pfama-1 sequences with the 3D7
pfama-1 reference sequence (U65407) revealed genetic polymorphisms in KP-Pakistan sequences. Across the sequences, 69 single nucleotide polymorphisms (SNPs) were identified, among which 58 were nonsynonymous SNPs (nsSNPs), resulting in amino acid substitutions at 58 positions and 14 distinct haplotypes. Most amino acid changes were found in DI (
n=24), DII (
n=7), and DIII (
n=6) (
Fig. 1;
Supplementary Table S2). A tetramorphic change (E197D/G/H) and 3 trimorphic changes (E187N/K, H200R/D, and K243E/N) were detected in DI. A trimorphic amino acid change (R503N/H) was identified in DIII. The other 53 amino acid changes throughout the sequences were dimorphic. We also comparatively analyzed KP-Pakistan
pfama-1 with previously reported global
pfama-1 and identified 113 nsSNPs causing amino acid substitutions (92 dimorphic, 17 trimorphic, 2 tetramorphic, and 2 pentamorphic) in global
pfama-1, including KP-Pakistan
pfama-1. Interestingly, 13 amino acid changes (W28D, H30D, R45S, K49T, Q57L, S66P, I97N, and M114V in the signal and prosequence region; D333E and N401S in DII, I454T in DIII; and K570K and P614S in the transmembrane and cytoplasmic domain) detected in KP-Pakistan
pfama-1 were novel that were not reported previously, although their frequencies were relatively low (
Fig. 1;
Supplementary Table S2). Meanwhile, the most amino acid changes in DI, DII, and DIII were commonly detected in global
pfama-1 (
Supplementary Table S2). Global
pfama-1 exhibited similar patterns of amino acid changes, but the frequency of each amino acid change differed by country.
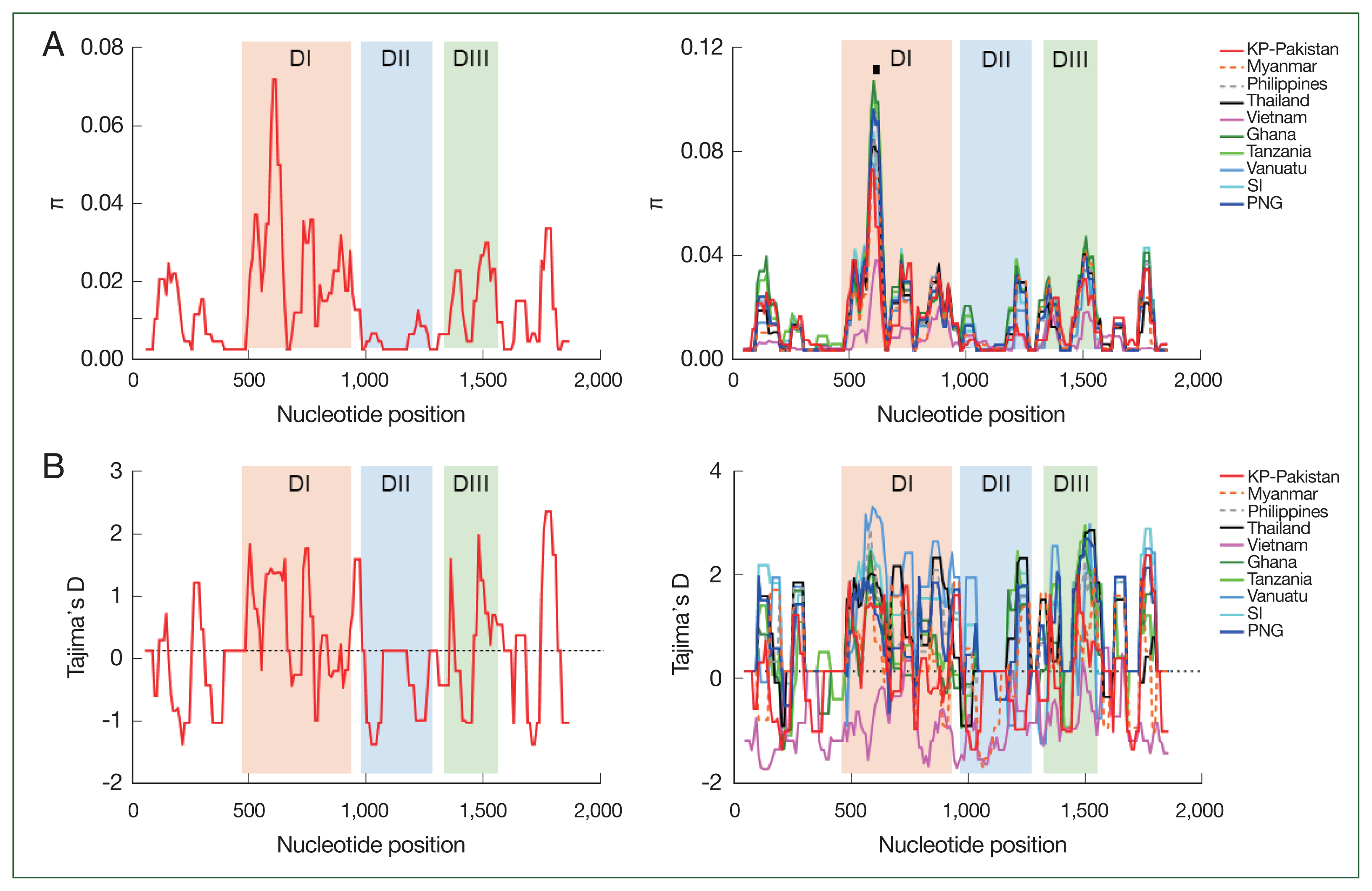
We identified 69 segregating sites and 69 mutations in KP-Pakistan
pfama-1 isolates. Haplotype diversity and nucleotide diversity (π) were 0.982±0.022 and 0.0109, respectively (
Table 1). Tajima’s D, Fu and Li’s D, and Fu and Li’s F values were positive, suggesting that positive natural selection affected KP-Pakistan
pfama-1. The overall nucleotide diversity (π) across global
pfama-1 ranged from 0.0043±0.0006 (Vietnam) to 0.0141±0.0003 (Ghana), suggesting mild levels of genetic diversity in global
pfama-1 populations. The π in KP-Pakistan
pfama-1 was lower than or similar to that in Asia and Pacific
pfama-1 populations but lower than that in Africa
pfama-1 populations (
Table 1). All
pfama-1 sequences, except Vietnam
pfama-1 sequences, demonstrated positive Tajima’s D values, indicating the role of balancing selection in global
pfama-1 (
Table 1). The positive values of both Fu and Li’s D and F also suggested evidence for the role of balancing selection in global
pfama-1, except Vietnam
pfama-1. A sliding window plot of π suggested that global
pfama-1 shared highly similar patterns of π across the sequences (
Fig. 2). The highest peak of π was commonly identified at cluster 1 of the loop I (C1-L) region in DI of all global isolates. Similar profiles of Tajima’s D across the gene were also identified in global
pfama-1, except Vietnam
pfama-1 (
Fig. 2). The
FST analysis between KP-Pakistan and global
pfama-1 populations indicated genetic differentiation of KP-Pakistan isolates. KP-Pakistan
pfama-1 showed the lowest
FST values against
pfama-1 from the Philippines, PNG and SI, but it showed higher
FST values against
pfama-1 from Myanmar, Thailand, and Vietnam (
Table 2).
The Rm of KP-Pakistan
pfama-1 was estimated to be 9. The values between adjacent sites (Ra) and per gene (Rb) were 0.0238 and 44.4, respectively (
Table 3). Possible recombination events were also identified in global
pfama-1. The Rm values of Africa
pfama-1 were greater than those of Asia and Pacific
pfama-1. The increasing distance across the gene with the decreased linkage disequilibrium index (R
2) in global
pfama-1 suggests that recombination could be a major force contributing to the genetic diversity of
pfama-1 (
Supplementary Fig. S2).
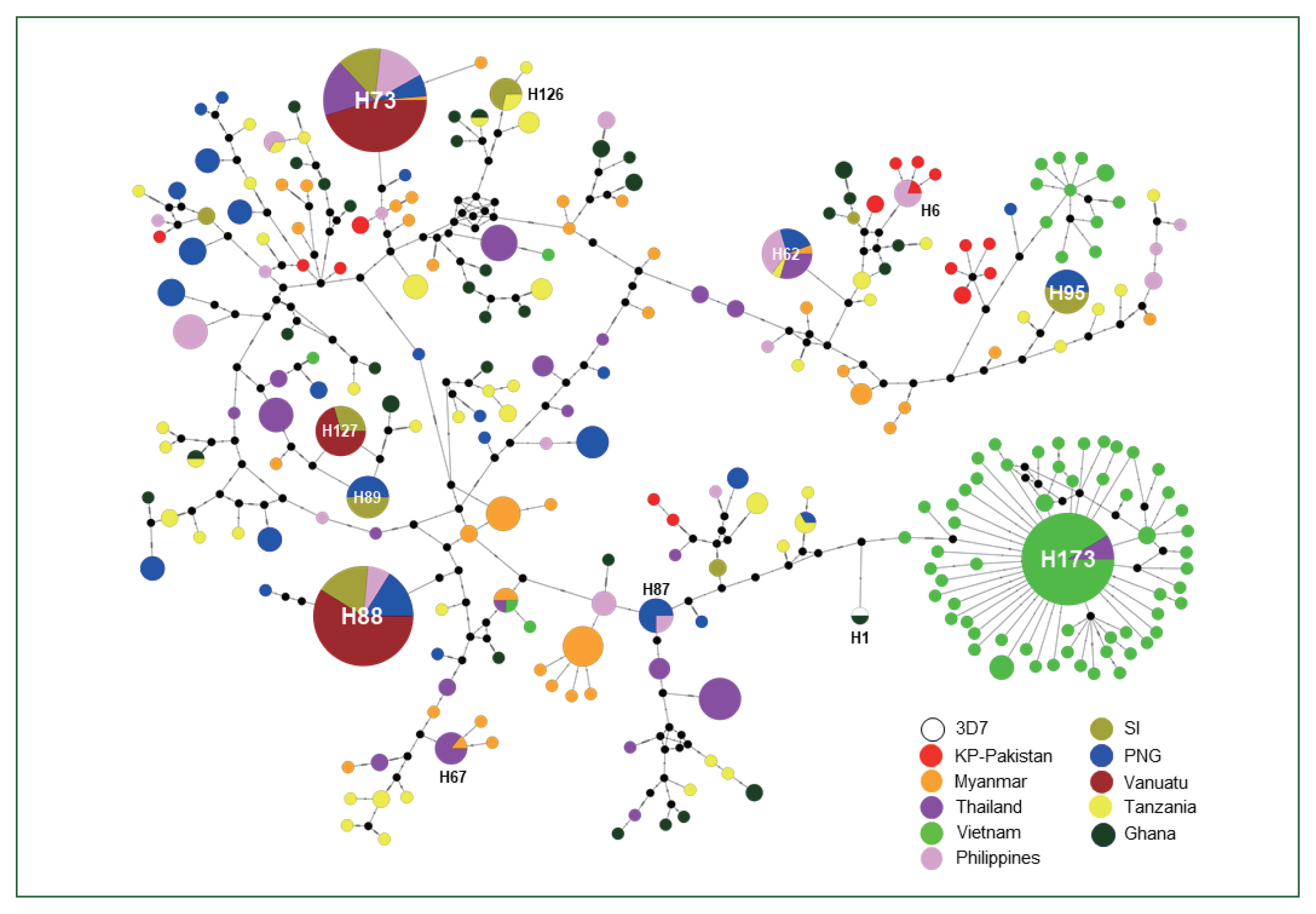
The haplotype network analysis of 667 global
pfama-1 and 3D7 reference sequences revealed a complicated network of 260 distinct haplotypes (
Fig. 3). Most haplotypes were singletons. Haplotype 73 (H73) was the most predominant with a frequency of 10.6% and was shared by
pfama-1 from different countries, including Myanmar, Thailand, the Philippines, Vanuatu, PNG, and SI. Haplotype 88 (H88) was the second major haplotype with a frequency of 10.2% and was shared by
pfama-1 from Vanuatu, SI, PNG, and the Philippines. KP-Pakistan
pfama-1 constructed 16 haplotypes that were scattered in the network. Only one sequence from Ghana shared a haplotype (H1) with 3D7.
We analyzed the pattern of natural selection signatures across global
pfama-1. The episodic positive selection analysis using the mixed effects model of evolution method suggested that 18 amino acid changes were under natural selection (
P≤0.05) (
Table 4). The pervasive positive selection signatures analyzed using fixed effects likelihood and internal branches fixed effects likelihood methods suggested that 20 amino acid changes were under natural selection (
Table 4). All amino acid changes predicted to be under positive natural selection matched the amino acid changes commonly detected in global
pfama-1.
Discussion
The complex biological properties of parasites and vectors and the large genetic and antigenic variations in prospective vaccine candidate antigens have hindered the development of an effective malaria vaccine despite extensive attempts over the past few decades. Although the RTS, S/A01, the first malaria vaccine endorsed by the World Health Organization for routine immunization of children in transmission areas, has developed based on CSP [
29], there are controversies on its efficacy due to modest and short-lived protection efficiency and insufficient effectiveness against parasites with different alleles of CSP [
30,
31]. To address these limitations, a multistage vaccine combining different candidate antigens such as Duffy binding protein and AMA-1 was proposed [
32,
33]. The biological significance and immunological functions of PfAMA-1 suggest that this antigen is an attractive vaccine candidate. Nonetheless, the genetic polymorphisms observed in global
pfama-1 also emphasize the importance of continuous surveillance of the genetic diversity of the gene in the global parasite population [
18,
19].
Similar to that in
pfama-1 from other geographical areas [
18,
19], KP-Pakistan
pfama-1 also exhibited genetic polymorphisms causing amino acid changes. The most common amino acid changes in global
pfama-1, especially those in DI, DII, and DIII, were also identified in KP-Pakistan
pfama-1. These common amino acid changes observed in DI and DIII of global
pfama-1 matched with B-cell epitopes 3, 4, 5, 9, and 10, supporting the notion that these are major regions under natural selection and contribute to host immune escape [
18,
19,
34]. Meanwhile, 13 novel amino acid changes not reported in global
pfama-1 were identified in KP-Pakistan
pfama-1, most of which were distributed in the signal and prosequence region. Among these amino acid changes, Q57L, S66P, I97N, I454T, and K570R were located in the intrinsically unstructured/disordered region, and D333E was mapped in red blood cell-binding sites. The amino acid changes I97N, M114V, D333E, N401S, and I454T were also located in the intrinsically unstructured/disordered region or B-cell epitope. These findings suggest the potential roles of these amino acids in the modulation of host immune responses; however, further studies would be required to clarify the biological significance of these amino acid changes.
KP-Pakistan
pfama-1 demonstrated similar patterns of genetic diversity and natural selection with those of
pfama-1 from other geographical regions. The π value for the global
pfama-1 population varied, with the π values of Asia and Pacific
pfama-1 populations being relatively lower than that of the Africa
pfama-1 population. Although the π value of global
pfama-1 differed by country, similar patterns of π across
pfama-1 were identified in global
pfama-1, including KP-Pakistan
pfama-1. The sliding window plot revealed that the high levels of π were similarly observed in DI and DIII of global
pfama-1, supporting that the domains are the central regions contributing to the genetic heterogeneity of
pfama-1 [
18,
19]. The positive values of Tajima’s D, Fu and Li’s D, and F of KP-Pakistan
pfama-1 suggested that the gene was under balancing selection. Similar patterns of natural selection were also detected in global
pfama-1, except Vietnam
pfama-1 [
18,
19,
35]. Meiotic recombination is also a driving force generating the genetic diversity of
pfama-1 [
18,
19]. Potential recombination events were also detected in KP-Pakistan
pfama-1, suggesting that interallelic recombination is a force causing the genetic diversity of KP-Pakistan
pfama-1.
FST is a measure of population substructure and is the most common statistic to analyze the overall genetic differentiation among populations as follows: no differentiation (0), low genetic differentiation (0–0.05), moderate differentiation (0.05–0.15), or high differentiation (0.15–0.25) [
36]. Global
pfama-1 displayed low or moderate levels of
FST between and among populations originating from different continents or countries. The only exception was Vietnam
pfama-1 [
19]. Although global
pfama-1 demonstrated a complicated haplotype diversity with 260 distinct haplotypes, low or moderate levels of
FST values between and among populations suggest that global
pfama-1 has a relatively stable genetic structure in the global population.
This study has some drawbacks due to the limited number of global pfama-1 sequences obtained from parasites collected at different time points, which could not reflect the genetic structure and evolutionary aspect of the current global pfama-1 population. Further in-depth analysis using a greater significant number of global P. falciparum populations is required to understand the genetic structure of pfama-1.
Notes
-
Author contributions
Conceptualization: Na BK, Afridi SG
Data curation: Zaib K, Khan A, Khan MU, Ullah I
Formal analysis: Zaib K, Khan A, Khan MU, Ullah I, Võ TC, Kang JM, Lê HG, Na BK, Afridi SG
Funding acquisition: Na BK
Investigation: Khan A, Na BK, Afridi SG
Methodology: Zaib K, Khan A, Khan MU, Ullah I, Võ TC, Kang JM
Project administration: Na BK, Afridi SG
Resources: Afridi SG
Software: Zaib K, Khan A, Khan MU, Ullah I, Võ TC, Kang JM, Lê HG, Na BK, Afridi SG
Supervision: Na BK, Afridi SG
Writing – original draft: Zaib K, Khan A, Afridi SG
Writing – review & editing: Khan A, Võ TC, Kang JM, Lê HG, Na BK, Afridi SG
-
The authors declare no conflicts of interests.
Supplementary Information
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant (NRF-2024M3A9H5043141).
Fig. 1Amino acid changes identified in KP-Pakistan PfAMA-1. The nsSNP-induced amino acid changes at 58 positions in the KP-Pakistan population compared with the reference 3D7 sequence (U65407). A total of 14 haplotypes were detected. A tetramorphic amino acid change (E197D/G/H) is marked with red letters. Trimorphic amino acid changes in DI and DIII are marked with blue letters. Asterisks indicated novel amino acid changes detected in KP-Pakistan pfama-1. DI, domain I; DII, domain II; DIII, domain III. Asterisks indicated novel amino acid changes detected in KP-Pakistan pfama-1.

Fig. 2Nucleotide diversity and natural selection of pfama-1. (A) Nucleotide diversity. The sliding window plot showed nucleotide diversity (π) values across KP-Pakistan pfama-1 sequences (left) and global pfama-1 sequences (right). Cluster 1 of the loop I (C1-L) region showing the highest π peak is marked with a black line. A window size of 100 bp and a step size of 25 bp were applied. (B) Natural selection. The sliding window plot of Tajima’s D was analyzed for KP-Pakistan pfama-1 sequences (left) and global pfama-1 sequences (right). A window size of 100 and a step size of 25 were applied. DI, domain I; DII, domain II; DIII, domain III.

Fig. 3Haplotype network plot. A total of 667 global pfama-1 sequences were analyzed. A circle represents each haplotype, and the circle size is proportional to the number of individual sequences corresponding to each haplotype. The length of lines connecting the haplotypes reflects the distance of relatedness. The color of each node indicates each country.

Table 1The estimates of DNA sequence polymorphism and tests of neutrality in pfama-1 among KP-Pakistan and global pfama-1
Table 1
|
Isolates |
Segregating sites (S) |
Singleton variable sites |
Parsimony informative sites |
Total no. of mutations |
K
|
H |
Hd±SD |
π±SD |
Tajima’s D |
Fu and Li’s D |
Fu and Li’s F |
|
KP-Pakistan (n=19) |
69 |
18 |
51 |
69 |
20.409 |
16 |
0.982±0.022 |
0.0109±0.0022 |
0.1391 (P>0.1) |
0.2370 (P>0.1) |
0.2420 (P>0.1) |
|
Vietnama (n=131) |
114 |
47 |
67 |
116 |
8.062 |
73 |
0.837±0.034 |
0.0043±0.0006 |
−2.0046 (P<0.05) |
−3.1913 (P<0.05) |
−3.1959 (P<0.05) |
|
Myanmarb (n=58) |
78 |
16 |
62 |
79 |
19.886 |
37 |
0.948±0.019 |
0.0106±0.0005 |
0.5719 (P>0.1) |
0.1677 (P>0.1) |
0.3851 (P>0.1) |
|
Thailandb (n=80) |
58 |
5 |
53 |
61 |
20.621 |
21 |
0.920±0.013 |
0.0110±0.0002 |
2.2222 (P<0.05) |
0.9572 (P>0.1) |
1.7516 (P<0.05) |
|
Philippinesb (n=55) |
61 |
3 |
58 |
62 |
21.232 |
19 |
0.916±0.019 |
0.0114±0.0003 |
1.9545 (0.05<P<0.1) |
1.6123 (P<0.05) |
2.0796 (P<0.05) |
|
PNGb (n=90) |
68 |
2 |
66 |
75 |
22.923 |
28 |
0.954±0.007 |
0.0123±0.0002 |
1.8082 (0.05<P<0.1) |
1.6759 (P<0.05) |
2.0760 (P<0.05) |
|
SIb (n=50) |
56 |
3 |
53 |
57 |
22.044 |
9 |
0.862±0.021 |
0.0118±0.0003 |
2.5455 (P<0.05) |
1.5447 (P<0.05) |
2.2804 (P<0.05) |
|
Vanuatub (n=85) |
50 |
6 |
44 |
50 |
18.905 |
5 |
0.633±0.028 |
0.0101±0.0003 |
2.8973 (P<0.1) |
0.8642 (P>0.1) |
2.0039 (P<0.05) |
|
Ghanab (n=37) |
75 |
7 |
68 |
85 |
26.261 |
32 |
0.992±0.008 |
0.0141±0.0003 |
1.0674 (P>0.1) |
1.2179 (P>0.1) |
1.3850 (P>0.1) |
|
Tanzaniab (n=62) |
81 |
8 |
73 |
89 |
25.444 |
47 |
0.989±0.005 |
0.0136±0.0002 |
1.1819 (P>0.1) |
1.1635 (P>0.1) |
1.4020 (P>0.1) |
Table 2Estimation of genetic differentiation between global pfama-1 population
Table 2
|
KP-Pakistan |
Myanmar |
Thailand |
Vietnam |
Philippines |
PNG |
SI |
Vanuatu |
Ghana |
Tanzania |
|
KP-Pakistan |
- |
|
|
|
|
|
|
|
|
|
|
Myanmar |
0.19025 |
- |
|
|
|
|
|
|
|
|
|
Thailand |
0.13256 |
0.06743 |
- |
|
|
|
|
|
|
|
|
Vietnam |
0.57838 |
0.45520 |
0.40917 |
- |
|
|
|
|
|
|
|
Philippines |
0.07009 |
0.09227 |
0.04578 |
0.46879 |
- |
|
|
|
|
|
|
PNG |
0.08690 |
0.05991 |
0.04382 |
0.40086 |
0.02891 |
- |
|
|
|
|
|
SI |
0.10522 |
0.10515 |
0.07468 |
0.48316 |
0.04465 |
0.02829 |
- |
|
|
|
|
Vanuatu |
0.18820 |
0.10437 |
0.11096 |
0.43044 |
0.07502 |
0.06868 |
0.05845 |
- |
|
|
|
Ghana |
0.11194 |
0.09206 |
0.05441 |
0.47405 |
0.05433 |
0.04658 |
0.06201 |
0.14206 |
- |
|
|
Tanzania |
0.11193 |
0.08872 |
0.06651 |
0.45434 |
0.06556 |
0.04287 |
0.06663 |
0.15077 |
0.01126 |
- |
Table 3Comparison of recombination events among global pfama-1
Table 3
|
n
|
Raa
|
Rbb
|
Rmc
|
|
KP-Pakistan |
19 |
0.0238 |
44.4 |
9 |
|
Vietnam |
131 |
0.0000 |
0.001 |
20 |
|
Myanmar |
78 |
0.0141 |
26.3 |
24 |
|
Thailand |
38 |
0.0243 |
45.3 |
20 |
|
Philippines |
61 |
0.0218 |
40.8 |
17 |
|
PNGd
|
68 |
0.0500 |
93.4 |
28 |
|
SIe
|
56 |
0.0120 |
22.5 |
14 |
|
Vanuatu |
50 |
0.0010 |
1.8 |
9 |
|
Ghana |
75 |
0.0921 |
172 |
27 |
|
Tanzania |
81 |
0.0915 |
171 |
28 |
Table 4The pfama-1 codons predicted to underline natural selection in KP-Pakistan and global samples
Table 4
|
Domain |
Amino acid position |
Episodic positive selection by MEME |
Pervasive positive selection |
|
|
FEL |
IFEL |
|
Signal and prosequence |
34a
|
|
* |
* |
|
35 |
|
* |
|
|
52a
|
|
* |
* |
|
|
DI |
167a
|
T-K |
* |
* |
|
172a
|
G-E |
* |
* |
|
187a
|
|
* |
* |
|
197a
|
E-Q/G/D/R/H |
* |
* |
|
200a
|
H-D/L/R |
|
|
|
242a
|
D-Y |
* |
* |
|
267a
|
|
* |
* |
|
283a
|
S-L |
* |
* |
|
300a
|
|
* |
|
|
308a
|
Q-E/K |
* |
* |
|
|
DII |
330a
|
P-S |
* |
* |
|
395 |
K-R |
* |
* |
|
404a
|
T-R |
* |
|
|
405a
|
|
* |
|
|
435a
|
I-N/T |
* |
* |
|
439a
|
N-H/D |
* |
* |
|
|
DIII |
451a
|
M-K |
* |
* |
|
485a
|
K-I |
* |
* |
|
493a
|
D-A |
* |
* |
|
503a
|
R-N/H |
* |
* |
|
|
Transmembrane and cytoplasmic |
512a
|
R-K |
* |
* |
|
544a
|
K-N |
* |
* |
References
- 1. World Health Organization. World Malaria Report 2022; World Health Organization; Geneva, Switzerland: 2022.
https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022
- 2. Richards JS, Beeson JG. The future for blood-stage vaccines against malaria. Immunol Cell Biol 2009;87(5):377-390.
https://doi.org/10.1038/icb.2009.27
- 3. Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature 2002;419(6906):520-526.
https://doi.org/10.1038/nature01107
- 4. Takala SL, Coulibaly D, Thera MA, Batchelor AH, Cummings MP, et al. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: implications for vaccine development. Sci Transl Med 2009;1(2):2ra5.
https://doi.org/10.1126/scitranslmed.3000257
- 5. Escalante AA, Lal AA, Ayala FJ. Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum
. Genetics 1998;149(1):189-202.
https://doi.org/10.1093/genetics/149.1.189
- 6. Healer J, Crawford S, Ralph S, McFadden G, Cowman AF. Independent translocation of two micronemal proteins in developing Plasmodium falciparum merozoites. Infect Immun 2002;70(10):5751-5758.
https://doi.org/10.1128/IAI.70.10.5751-5758.2002
- 7. Silvie O, Franetich JF, Charrin S, Mueller MS, Siau A, et al. A role for apical membrane antigen 1 during invasion of hepatocytes by Plasmodium falciparum sporozoites. J Biol Chem 2004;279(10):9490-9496.
https://doi.org/10.1074/jbc.M311331200
- 8. Hodder AN, Crewther PE, Matthew ML, Reid GE, Moritz RL, et al. The disulfide bond structure of Plasmodium apical membraneantigen-1. J Biol Chem 1996;271:29446-29452.
https://doi.org/10.1074/jbc.271.46.29446
- 9. Udhayakumar V, Kariuki S, Kolczack M, Girma M, Roberts JM, et al. Longitudinal study of natural immune responses to the Plasmodium falciparum apical membrane antigen (AMA-1) in a holoendemic region of malaria in western Kenya: Asembo Bay Cohort Project VIII. Am J Trop Med Hyg 2001;65(2):100-107.
https://doi.org/10.4269/ajtmh.2001.65.100
- 10. Moncunill G, Aponte JJ, Nhabomba AJ, Dobano C. Performance of multiplex commercial kits to quantify cytokine and chemokine responses in culture supernatants from Plasmodium falciparum stimulations. PLoS One 2013;8(1):e52587.
https://doi.org/10.1371/journal.pone.0052587
- 11. Rodrigues MH, Rodrigues KM, Oliveira TR, Cômodo AN, Rodrigues MM, et al. Antibody response of naturally infected individuals to recombinant Plasmodium vivax apical membrane antigen-1. Int J Parasitol 2005;35(2):185-192.
https://doi.org/10.1016/j.ijpara.2004.11.003
- 12. Gentil EC, Damgaard A, Hauschild M, Finnveden G, Eriksson O, et al. Models for waste life cycle assessment: review of technical assumptions. Waste Manag 2010;30(12):2636-2648.
https://doi.org/10.1016/j.wasman.2010.06.004
- 13. Remarque EJ, Faber BW, Kocken CH, Thomas AW. Apical membrane antigen 1: a malaria vaccine candidate in review. Trends Parasitol 2008;24(2):74-84.
https://doi.org/10.1016/j.pt.2007.12.002
- 14. Thera MA, Coulibaly D, Kone AK, Guindo AB, Traore K, et al. Phase 1 randomized controlled trial to evaluate the safety and immunogenicity of recombinant Pichia pastoris-expressed Plasmodium falciparum apical membrane antigen 1 (PfAMA1-FVO [25–545]) in healthy Malian adults in Bandiagara. Malar J 2016;15(1):442.
https://doi.org/10.1186/s12936-016-1466-4
- 15. Volkman SK, Neafsey DE, Schaffner SF, Park DJ, Wirth DF. Harnessing genomics and genome biology to understand malaria biology. Nat Rev Genet 2012;13(5):315-328.
https://doi.org/10.1038/nrg3187
- 16. Khan MI, Qureshi H, Bae SJ, Khattak AA, Anwar MS, et al. Malaria prevalence in Pakistan: a systematic review and meta-analysis (2006–2021). Heliyon 2023;9(4):e15373.
https://doi.org/10.1016/j.heliyon.2023.e15373
- 17. Afridi SG, Irfan M, Ahmad H, Aslam M, Nawaz M, et al. Population genetic structure of domain I of apical membrane antigen-1 in Plasmodium falciparum isolates from Hazara division of Pakistan. Malar J 2018;17(1):389.
https://doi.org/10.1186/s12936-018-2539-3
- 18. Kang JM, Lee J, Moe M, Jun H, Lê HG, et al. Population genetic structure and natural selection of Plasmodium falciparum apical membrane antigen-1 in Myanmar isolates. Malar J 2018;17(1):71.
https://doi.org/10.1186/s12936-018-2215-7
- 19. Kang JM, Lê HG, Võ TC, Naw H, Yoo WG, et al. Genetic polymorphism and natural selection of apical membrane antigen-1 in Plasmodium falciparum isolates from Vietnam. Genes (Basel) 2021;12(12):1903.
https://doi.org/10.3390/genes12121903
- 20. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 2013;30(12):2725-2729.
https://doi.org/10.1093/molbev/mst197
- 21. Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 2017;34(12):3299-3302.
https://doi.org/10.1093/molbev/msx248
- 22. Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 2005;1(4):47-50.
https://doi.org/10.1111/j.1755-0998.2010.02847.x
- 23. Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 1999;16(1):37-48.
https://doi.org/10.1093/oxfordjournals.molbev.a026036
- 24. Penn O, Privman E, Ashkenazy H, Landan G, Graur D, et al. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res 2010;38(Web Server issue):23-28.
https://doi.org/10.1093/nar/gkq443
- 25. Privman E, Penn O, Pupko T. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol biol and Evol 2012;29(1):1-5.
https://doi.org/10.1093/molbev/msr177
- 26. Kosakovsky Pond SL, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005;21(10):2531-2533.
https://doi.org/10.1093/bioinformatics/bti320
- 27. Delport W, Poon AFY, Frost SDW, Kosakobsky Pond SL. Kosakovsky Pond, Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010;26(19):2455-2457.
https://doi.org/10.1093/bioinformatics/btq429
- 28. Murrell B, Wertheim JO, Moola S, Weighill T, Scheffler K, et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 2012;8(7):1-10.
https://doi.org/10.1371/journal.pgen.1002764
- 29. RTSS Clinical Trials Partnership, Agnandji ST, Lell B, Soulanoudjingar SS, Fernandes JF, et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N Engl J Med 2011;365(20):1863-1875.
https://doi.org/10.1056/NEJMoa1102287
- 30. Neafsey DE, Juraska M, Bedford T, Benkeser D, Valim C, et al. Genetic diversity and protective efficacy of the RTS,S/AS01 malaria vaccine. N Engl J Med 2015;373(21):2025-2037.
https://doi.org/10.1056/NEJMoa1505819
- 31. Beeson JG, Kurtovic L, Valim C, Asante KP, Boyle MJ, et al. The RTS, S malaria vaccine: current impact and foundation for the future. Sci Transl Med; 2022. 14(671):eabo6646
https://doi.org/10.1126/scitranslmed.abo6646
- 32. Boes A, Spiegel H, Kastilan R, Bethke S, Voepel N, et al. Analysis of the dose-dependent stage-specific in vitro efficacy of a multi-stage malaria vaccine candidate cocktail. Malar J 2016;15(1):279.
https://doi.org/10.1186/s12936-016-1328-0
- 33. Rajneesh , Tiwari R, Singh VK, Kumar A, Gupta RP, et al. Advancements and challenges in developing malaria vaccines: targeting multiple stages of the parasite life cycle. ACS Infect Dis 2023;9(10):1795-1814.
https://doi.org/10.1021/acsinfecdis.3c00332
- 34. Bai T, Becker M, Gupta A, Strike P, Murphy VJ, et al. Structure of AMA1 from Plasmodium falciparum reveals a clustering of polymorphisms that surround a conserved hydrophobic pocket. Proc Natl Acad Sci U S A 2005;102(36):12736-12741.
https://doi.org/10.1073/pnas.0501808102
- 35. Zhu X, Zhao Z, Feng Y, Li P, Liu F, et al. Genetic diversity of the Plasmodium falciparum apical membrane antigen I gene in parasite population from the China-Myanmar border area. Infect Genet Evol 2016;39:155-162.
https://doi.org/10.1016/j.meegid.2016.01.021
- 36. Balloux F, Lugon-Moulin N. The estimation of population differentiation with microsatellite markers. Mol Ecol 2002;11(2):155-165.

 , Asifullah Khan1,†, Muhammad Umair Khan1, Ibrar Ullah1, Tuấn Cường Võ2, Jung-Mi Kang2, Hương Giang Lê2
, Asifullah Khan1,†, Muhammad Umair Khan1, Ibrar Ullah1, Tuấn Cường Võ2, Jung-Mi Kang2, Hương Giang Lê2