Abstract
The gut microbiome plays an essential role in host immune responses, including allergic reactions. However, commensal gut microbiota is extremely sensitive to antibiotics and excessive usage can cause microbial dysbiosis. Herein, we investigated how changes in the gut microbiome induced by ampicillin affected the production of IgG1 and IgG2a antibodies in mice subsequently exposed to Anisakis pegreffii antigens. Ampicillin treatment caused a notable change in the gut microbiome as shown by changes in both alpha and beta diversity indexes. In a 1-dimensional immunoblot using Anisakis-specific anti-mouse IgG1, a 56-kDa band corresponding to an unnamed Anisakis protein was detected using mass spectrometry analysis only in ampicillin-treated mice. In the Anisakis-specific anti-mouse IgG2a-probed immunoblot, a 70-kDa band corresponding to heat shock protein 70 (HSP70) was only detected in ampicillin-treated and Anisakis-immunized mice. A 2-dimensional immunoblot against Anisakis extract with immunized mouse sera demonstrated altered spot patterns in both groups. Our results showed that ampicillin treatment altered the gut microbiome composition in mice, changing the immunization response to antigens from A. pegreffii. This research could serve as a basis for developing vaccines or allergy immunotherapies against parasitic infections.
-
Key words: Anisakis pegreffii, immunoglobulin, immunoblot, ampicillin, microbiome, allergen
Introduction
Anisakis spp. are nematode parasites that infect a wide range of marine organisms.
Anisakis also causes a human disease by consuming raw or undercooked fish infected with
Anisakis spp., such as
Anisakis simplex,
Anisakis pegreffii, and
Anisakis physeteris [
1]. The severity of the disease varies from mild to severe and presents with distinct pathological symptoms [
2]; however, parasite infection may cause adverse complications, such as abdominal pain, nausea, vomiting, allergy, anaphylactic shock, and even death [
3].
Anisakis is the only parasite known to act as a food allergen and is a major cause of food allergy, causing allergies more often than seafood itself [
3].
A. simplex and
A. pegreffii show a worldwide distribution and are the major causes of anisakiasis [
3]. Thus,
Anisakis spp. are potential seafood-borne pathogens and allergens for humans, posing severe health and financial impacts on fisheries industry, where fishermen and handlers are at risk of developing occupational asthma caused by the inhalation of allergens produced by
Anisakis spp. [
4].
The World Health Organization (WHO) in collaboration with the International Union of Immunological Societies (IUIS) has entrusted the WHO/IUIS Allergen Nomenclature Subcommittee with formulating a systematic and unequivocal nomenclature for allergenic proteins. Within this framework, 14
Anisakis allergens have been registered as Ani s 1–14 (
www.allergen.org). Ani s 1, 2, 3, 7, 12, and 13 are major allergens [
3], with Ani s 1, 4–9, and 13 being parasite excretory/secretory molecules; Ani s 2, 3, and 12 are somatic antigens [
5]; and Ani s 1, 4, 5, 8, and 9 are heat-stable allergens, and patients sensitized to these develop adverse allergic reactions to cooked or canned fish. Most of the heat-stable allergens are present in the excretory/secretory products of
Anisakis [
5]. Moreover, several allergens are resistant even to freezing or pepsin treatment [
5]. Kim et al. investigated the molecular and immunological aspects of allergen-like entities from parasites, emphasizing their role in allergic reactions and their diagnostic and therapeutic significance [
3].
The gut microbiome composition can be affected by age, diet, and environmental conditions [
6]. The gastrointestinal tract commensal microbiota are essential for health as they contribute to the maturation of the mucosal and systemic immune systems and aid in resisting colonization by invading pathogens [
6]. Furthermore, the gastrointestinal tract plays an important role in the development of either effector or tolerant responses to different antigens by balancing the activity of the T helper (Th) 1 and Th2 response [
7]. Perturbations that disrupt the healthy composition of the gut microbiome may contribute to immune dysfunction in allergic diseases, such as allergic rhinitis and asthma [
8]. Additionally, several studies have shown that antibiotics administered early in life significantly increase the risk of asthma in children [
9]. Antibiotics cause dysbiosis of gut microbiota, disrupting the balance between the populations of the Th1 and Th2 cells, which can favor an increase in the frequency of allergen-based reactions and consequently decrease the quality of life of an individual [
10]. Although gut microbial dysbiosis has been connected to the onset of food allergies, the intricate pathways involved remain under scrutiny. To decipher the comprehensive dynamics between gut dysbiosis and allergic manifestations, a more profound exploration is essential.
Herein, we investigated the effect of gut microbial dysbiosis on the synthesis of immunoglobulin (Ig) G1 and IgG2a antibodies on subsequent exposure to A. pegreffii protein extract. Importantly, IgG1 correlates with Th2 immune responses while IgG2a aligns with Th1 responses, both playing pivotal roles in allergic manifestations, although the production of IgE or other allergy indicators was not assessed in our study. To examine the changes in immunized allergen patterns caused by alterations in the composition of gut microbiota, we treated mice with ampicillin to eliminate gut microbiota. Proteomics on a 2-dimensional (2-D) immunoblot of A. pegreffii protein extract was performed using sera from mice immunized with A. pegreffii. Subsequently, we assessed whether each antigen was immunized with Th1-related IgG2a or Th2-related IgG1 and confirmed that changes in the microbiome could alter the immunization of individual proteins of A. pegreffii.
Materials and Methods
Ethics statement
All methodologies involving experimental animals received approval from the Department of Laboratory Animal Resources Committee of Yonsei University College of Medicine (no. 2020-0055).
Protein preparation
A single Chub mackerel (
Scomber japonicus) was acquired from the Saruga shopping center, Seoul, Korea;
Anisakis third-stage larvae were manually collected from the abdominal cavity, yielding a total of 150 larvae, which were washed before DNA and protein extraction processes. DNA from 15
Anisakis larvae was extracted using the NucleoSpin DNA Insect Kit (Macherey-Nagel, Düren, Germany). The presence of
A. pegreffii was confirmed through polymerase chain reaction (PCR) using primers targeting the ITS region: Primer A (forward: 5′-GTCGAATTCGTAGGTGAACCTGCGGAAGGATCA-3′) and Primer B (reverse: 5′-GCCGGATCCGAATCCTGGTTAGTTTCTTTTCCT-3′) [
11]. PCR amplifications were sequenced and aligned with
A. pegreffii reference sequences obtained from GenBank, confirming that the 15 tested parasites were
A. pegreffii. Protein extraction from the remaining 135 larvae was performed using sonication, followed by centrifugation at 10,000×g for 30 min and filtration through a sterilized 0.22-μm filter (Merck Millipore, Darmstadt, Germany). The protein concentration was determined using the Bradford assay (Bio-Rad, Hercules, CA, USA). The extract was maintained on ice throughout the extraction process and stored at −80°C.
Female BALB/c mice (
n=15; 6 weeks old) were purchased from Orient Bio (Seongnam, Korea) and tested in 3 groups (5 mice per group): phosphate-buffered saline (PBS) group,
Anisakis-immunized without ampicillin treatment group, and
Anisakis-immunized with ampicillin group; the number of mice per group was consistent with that used in our previous
Anisakis allergy research [
12]. To ensure optimum conditions, mice were housed in a specific pathogen-free environment with a 12-h light/dark cycle and given a week to acclimatize before experimentation. A daily health assessment was systematically conducted per mouse.
For immunization, 100 μg of
A. pegreffii protein extract was suspended in PBS while ampicillin was prepared at a concentration of 1 g/L in distilled water [
13]. This concentration was then provided to specific groups as part of their daily water consumption. The ampicillin regimen was commenced a week before the primary sensitization and maintained throughout the experiment. Mice were intraperitoneally injected with the
A. pegreffii extract (100 μg) once weekly for a total of 4 weeks [
14]. Mice were euthanized a fortnight after the final dosage. This study was repeated 3 times.
Mice were euthanized using CO
2 gas, and blood was collected from the inferior vena cava. For serum separation, blood samples were centrifuged for 30 min at 3,000×g, and the supernatant was collected. The serum levels of
A. pegreffii-specific IgG1 and IgG2a were measured using an enzyme-linked immunosorbent assay (ELISA). Briefly, 1×8 Stripwell 96-well plates (Corning, Corning, NY, USA) were coated with 100 μl/well coating buffer containing 0.3 μg
A. pegreffii protein extract and incubated overnight at 4°C [
12]. The plate was then washed with PBS+0.05% of Tween-20 (PBST) and blocked with 200 μl/well diluent buffer (PBST containing 1% of bovine serum albumin (BSA)) for 1 h at 25°C. Samples were diluted 1:100 with diluent buffer. After blocking, 100 μl of samples were dispensed per well and incubated for 2 h. Biotinylated goat anti-mouse IgG1 or biotin goat anti-mouse IgG2a (both diluted 1:1,000; NBP1-69914B and NBP1-69915B, respectively; Novus Biologicals, Littleton, CO, USA) were used as secondary antibodies. Wells were then incubated with an avidin-horseradish peroxidase (HRP) conjugate (BioLegend, San Diego, CA, USA) for 30 min, followed by incubation with 3,3,5,5-tetramethylbenzidine (Sigma-Aldrich) substrate (50 μl) in the dark for 5 min. The reaction was inhibited by adding 0.5M H
2SO
4. Absorbance was measured at 450 nm using VersaMax (Molecular Devices, Seoul, Korea). Statistical analyses were performed using GraphPad Prism software (version 8.0, GraphPad Software, San Diego, CA, USA).
Western blotting was performed using A. pegreffii protein. Proteins were separated by gel electrophoresis. A. pegreffii protein (10 μg) was loaded into each well of a mini format 10% sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel. Following gel electrophoresis, proteins were transferred from the gel onto a polyvinylidene difluoride membrane. After transfer, the membrane was blocked with 5 ml of Tris-buffered saline Tween-20 (TBST) containing 5% of BSA for 2 h at 25°C. Pooled samples were diluted 1:1,000 with TBST containing 1% of BSA and incubated overnight at 4°C. The goat anti-mouse IgG1 and goat anti-mouse IgG2a heavy chain antibodies (both diluted 1:1,000; ab97240 and ab97245, respectively; Abcam, Cambridge, UK) were used as secondary antibodies conjugated with HRP (1:10,000; ab2116, Abcam) for detection. Bands were developed using the ECL reaction (Bio-Rad), and images were captured using Chemidoc XRS (Bio-Rad).
Sample preparation and 2-D separation
Protein concentrations were determined using the Bradford method (Bio-Rad). For 2-D analysis, pH 3–10 IPG strips (GE Healthcare Life Sciences, Pittsburgh, PA, USA) were rehydrated in swelling buffer containing 7 M urea, 2 M thiourea, 2.5% (w/v) dithiothreitol, and 4% (w/v) CHAPS. Protein lysates (600 μg) were loaded into the rehydrated IPG strips using an IPGphor III (GE Healthcare Life Sciences), and the 2-D separation was performed on a 10% SDS-PAGE gel. Following fixation of the gels for 1 h in a solution of 40% (v/v) methanol containing 5% (v/v) phosphoric acid, gels were stained with colloidal Coomassie blue G-250 solution (ProteomeTech, Seoul, Korea). Gels were destained using deionized water, and images were acquired via an image scanner (Bio-Rad). Image analysis was performed using ImageMaster 2-D Platinum software (Amersham Biosciences, Hercules, CA, USA). To compare protein spots, >25 spots in all gels were marked and normalized. Data for the normalized spot intensities are shown in
Table 1.
Spots from 2-D gels were excised and in-gel digested with trypsin [
15]. Briefly, protein spots were excised from the stained gels and cut into pieces, which were washed for 1 h at 25°C in 25 mM ammonium bicarbonate buffer, pH 7.8, containing 50% (v/v) acetonitrile (ACN). Following dehydration of gel pieces in a centrifugal vacuum concentrator (Biotron, Inc., Incheon, Korea) for 10 min, they were rehydrated in 50 ng of sequencing-grade trypsin solution (Promega, Madison, WI, USA). After incubation in 25 mM ammonium bicarbonate buffer, pH 7.8, at 37°C overnight, tryptic peptides were extracted in 100 μl of 1% formic acid (FA) containing 50% (v/v) ACN for 20 min with mild sonication. The extracted solution was then concentrated using a centrifugal vacuum concentrator. Before mass spectrometry (MS) analysis, the peptide solution was desalted using a reversed-phase column [
16]. Briefly, after an equilibration step with 10 μl of 5% (v/v) FA, the peptide solution was loaded onto the column and washed with 10 μl of 5% (v/v) FA. Bound peptides were eluted in 8 μl of 70% ACN in 5% (v/v) FA.
Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis was performed using a nano ACQUITY UPLC and LTQ-orbitrap-mass spectrometer (Thermo Electron, San Jose, CA, USA) with a BEH C18 1.7 μm, 100 μm×100 mm column (Waters, Milford, MA, USA). The mobile phase A for the LC separation was 0.1% FA in deionized water, and the mobile phase B was 0.1% FA in ACN. The chromatography gradient provided a linear increase from 10% B to 40% B for 16 min, from 40% B to 95% B for 8 min, and from 90% B to 10% B for 11 min. The flow rate was 0.5 μl/min. For tandem MS, mass spectra were acquired using data-dependent acquisition with a full mass scan (300–2,000
m/z), followed by MS/MS scans. Each MS/MS scan acquired was the average of 1 microscan of the LTQ. The temperature of the ion transfer tube was controlled at 275°C, and the spray voltage was 2.3 kV. The normalized collision energy was set to 35% for MS/MS. Individual spectra from MS/MS were processed using the SEQUEST software (Thermo Quest, San Jose, CA, USA), and the generated peak lists were used to query the in-house database using the MASCOT program (Matrix Science, London, UK). The modifications of carbamidomethyl, deamidated, and oxidation were set for MS analysis, and the tolerance of the peptide mass was 10 ppm. The MS/MS ion mass tolerance was 0.8 Da, the allowance of missed cleavage was 2, and charge states +2 and +3 were considered for data analysis. We considered only significant hits as defined by the MASCOT probability analysis [
17].
Mouse ceca were collected and stored at −80°C. Cecal DNA was extracted using the FastDNA SPIN Kit for Soil (MP Biomedicals, Carlsbad, CA, USA). DNA corresponding to the V3–V4 region of the 16S rRNA gene was PCR amplified using the following bacterial universal primer pair: forward primer, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′, and reverse primer, 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′ [
18]. Limited-cycle amplification was performed using the Nextera XT Index Kit (Illumina, San Diego, CA, USA) to add multiplexing indices and Illumina sequencing adapters to PCR products. Libraries were normalized, pooled, and sequenced using the MiSeq platform (600 cycles, Illumina MiSeq V3 cartridge; Illumina).
Bioinformatics analyses were performed as described by Kim et al. [
18] Raw reads were processed through a quality check, and low-quality (Q<25) reads were filtered using Trimmomatic 0.32 [
19]. Paired-end sequence data were merged using PandaSeq [
20]. Primers were trimmed using the ChunLab in-house program (ChunLab, Inc., Seoul, Korea), applying a similarity cutoff threshold of 0.8. Sequences were denoised using the Mothur preclustering program, which merges and extracts unique sequences, allowing up to 2 differences between them [
21]. The EzBioCloud database [
22] was used for taxonomic assignment using BLAST 2.2.22 [
23], and pairwise alignments were generated to calculate the similarity [
24]. The UCHIME algorithm and nonchimeric 16S rRNA database from EzBioCloud were used to detect chimeric sequences for reads with the best hit similarity rate of <97% [
25]. Sequence data were then clustered using CD-Hit and UCLUST [
26,
27]. All aforementioned analyses were performed using EzBioCloud, a commercially available ChunLab bioinformatics cloud platform for microbiome research (
https://www.ezbiocloud.net/). Reads were normalized by random subsampling to 7,666 to perform the analyses. The Shannon index [
28] and principal coordinate analysis (PCoA) [
29] were computed based on the generalized UniFrac distance [
30]. The Kruskal–Wallis test was used to compare differences between the number of operational taxonomic units (OTUs), and the Shannon index was used to compare microbiome diversity among the 3 groups. Linear discriminant analysis effect size (LEfSe) analysis was used to identify significantly different taxa [
31].
Results
Metabarcoding of Anisakis-immunized mice
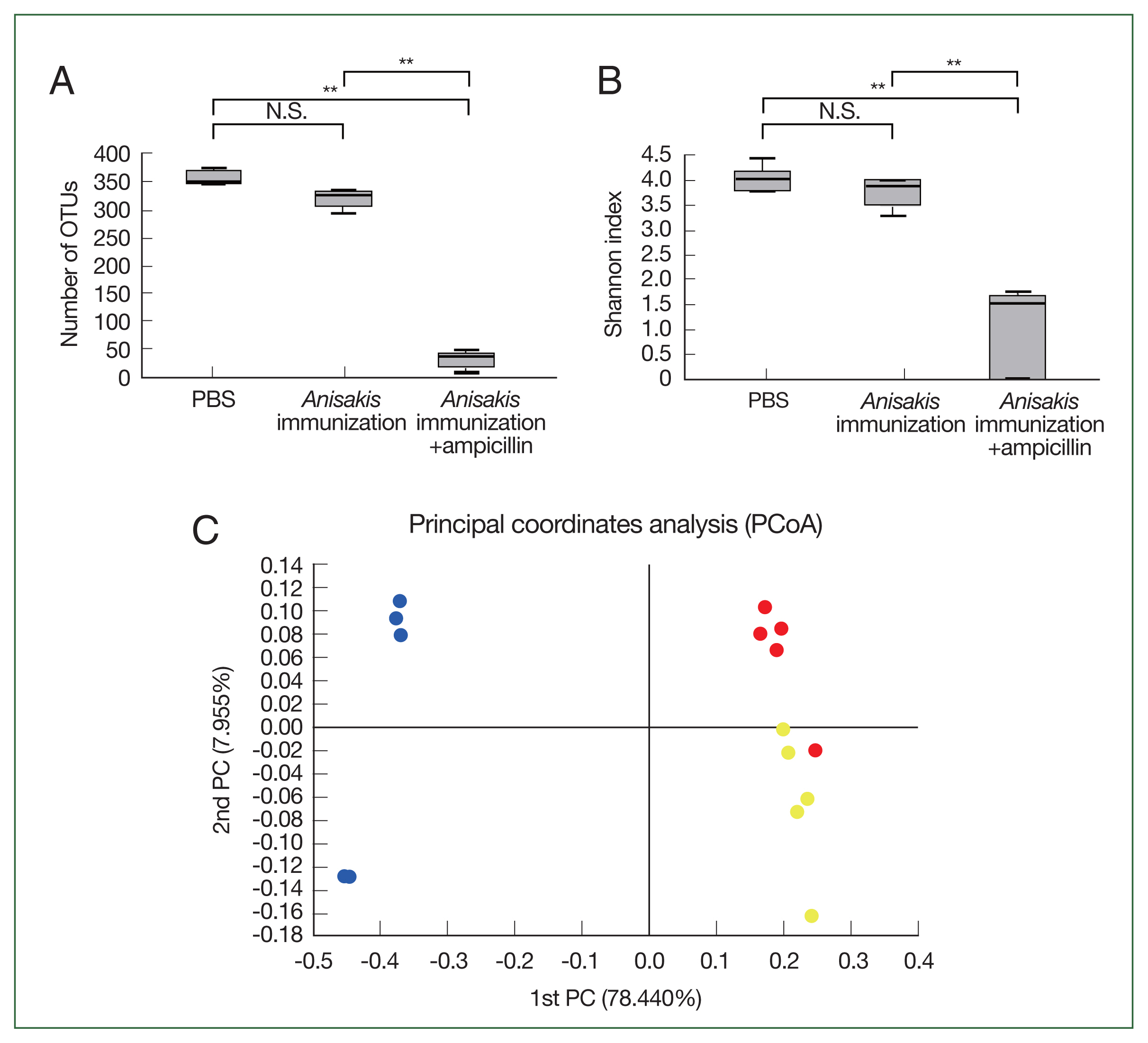
In the cecal microbiome study, the number of OTUs and Shannon index significantly decreased in the ampicillin-treated and
Anisakis-immunized mice (
Fig. 1A, B), indicating that the cecal microbiome diversity decreased after ampicillin treatment. In PCoA, the ampicillin-treated samples were distinctly clustered from untreated counterparts along the PCoA1 axis (
Fig. 1C). Composition analysis suggested that the ampicillin-treated group exhibited the pronounced presence of a bacterial species resembling
Proteus while the relative abundance of
Bacteroides was markedly different across the groups (
Fig. 2). We then performed LEfSe analysis to identify significant differences in bacterial abundance among the control and ampicillin-treated and untreated
Anisakis-immunized groups. The LDA score for
Proteus vulgaritus was 5.54, and the average relative abundance was 63.12%, 0.04%, and 0.02%, respectively in the ampicillin-treated
Anisakis-immunized, control, and ampicillin-untreated
Anisakis-immunized groups. The LDA score for
Bacteroides_uc was 5.16, and its average relative abundance was 21.92%, 28.28%, and 0.08% in the control, ampicillin-untreated
Anisakis-immunized, and ampicillin-treated
Anisakis-immunized groups, respectively (
Table 2).
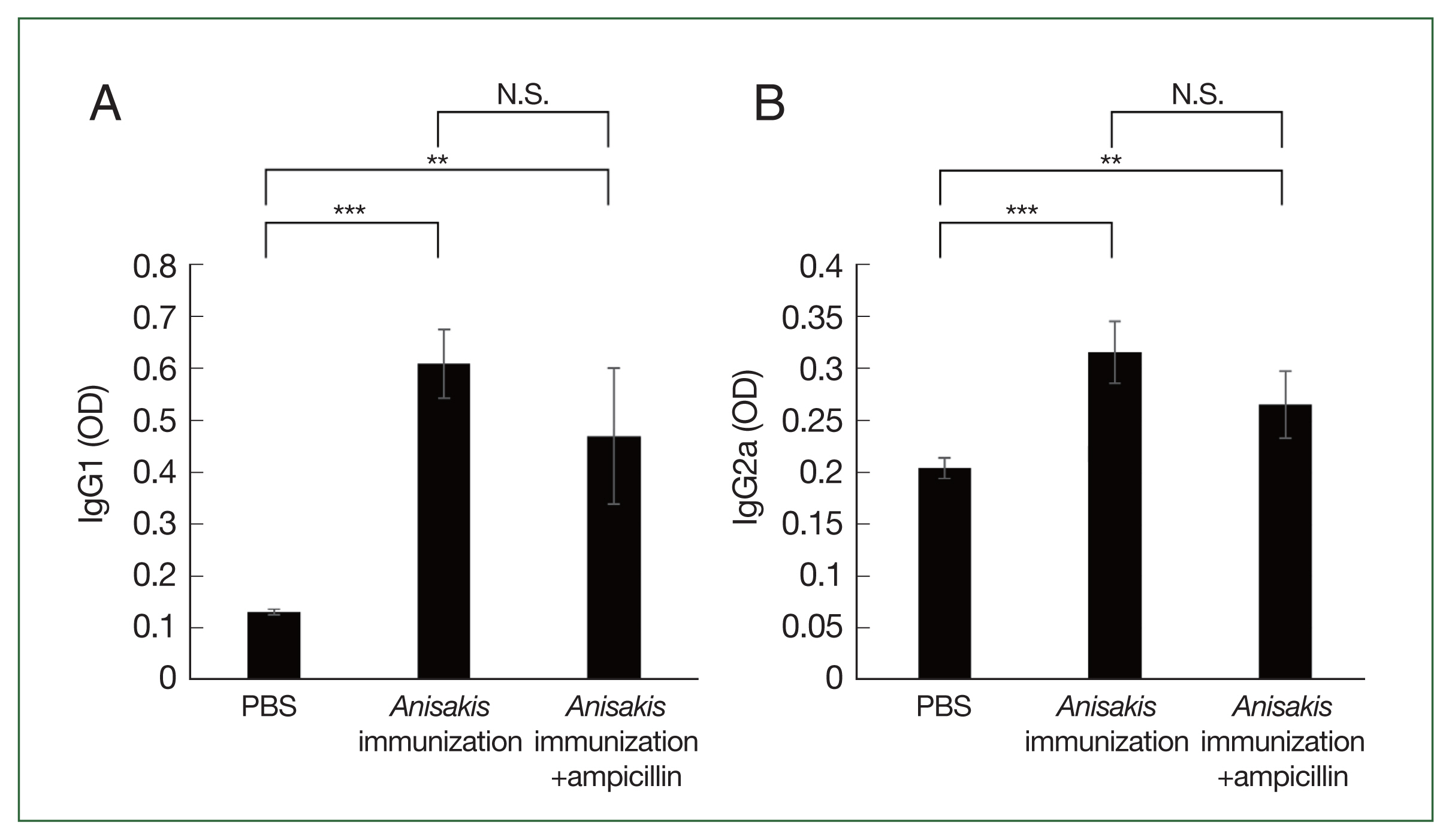
ELISA analysis showed that the
Anisakis-specific IgG1 and IgG2a levels increased in the
Anisakis-immunized mice with or without ampicillin treatment, although the difference in the IgG1 and IgG2a levels between
Anisakis-immunized ampicillin-treated and untreated groups was not significantly different (
Fig. 3A, B).
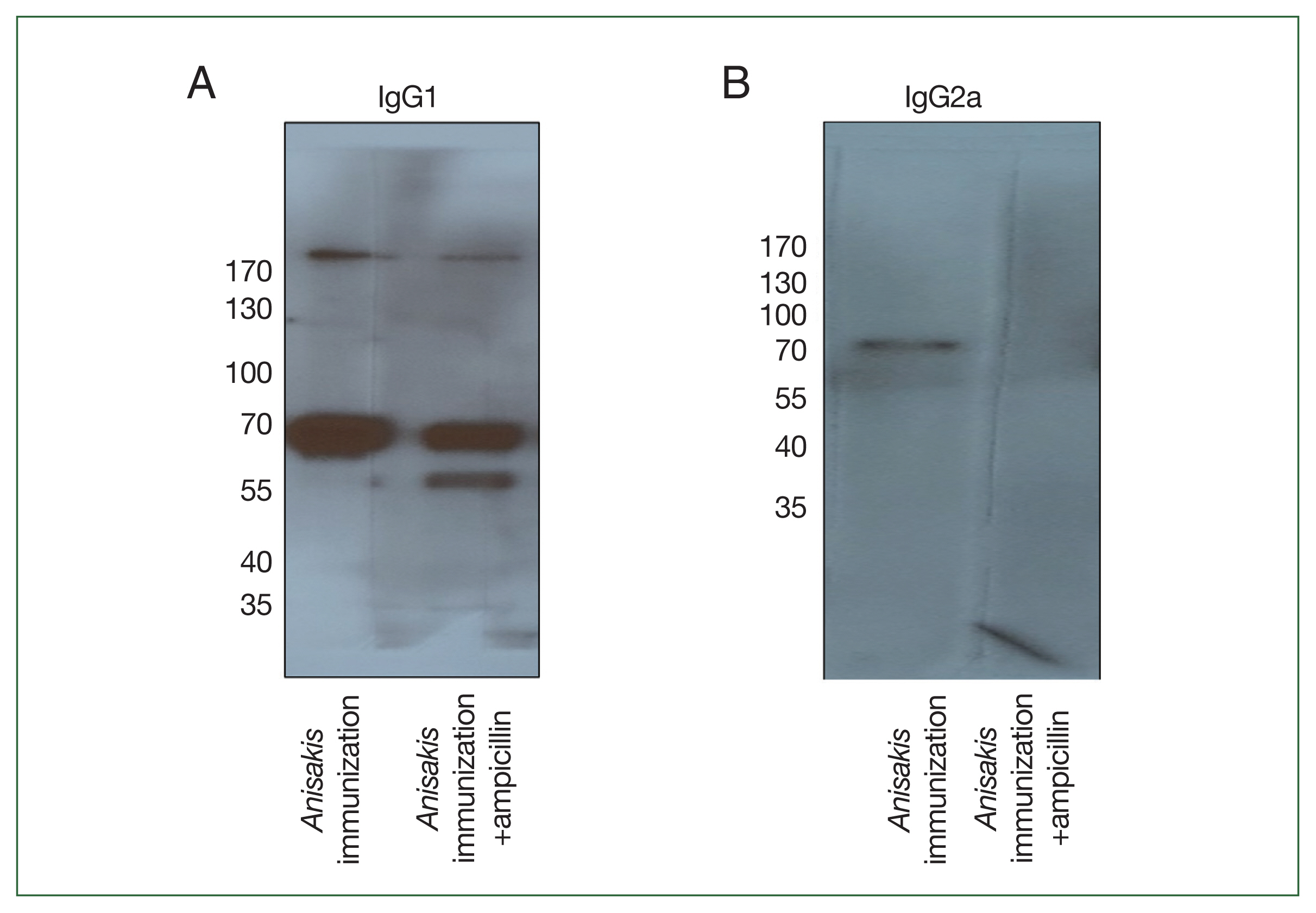
To determine which
Anisakis antigens were immunized in mice, immunoblotting was performed using pooled sera. In a 1-dimensional (1-D) immunoblot using
Anisakis-specific anti-mouse IgG1, a 56-kDa band was detected only in the ampicillin treatment group and identified as an “unnamed protein product (VDK17787.1 and VDK17834.1)” in MS analysis (
Fig. 4A;
Table 3). In an immunoblot with
Anisakis-specific anti-mouse IgG2a, a band at 70 kDa disappeared in the ampicillin treatment group and was identified as an unnamed protein product (VDK17973.1) and heat shock protein 70 (HSP70) (AIU38247.1) (
Fig. 4B;
Table 3).
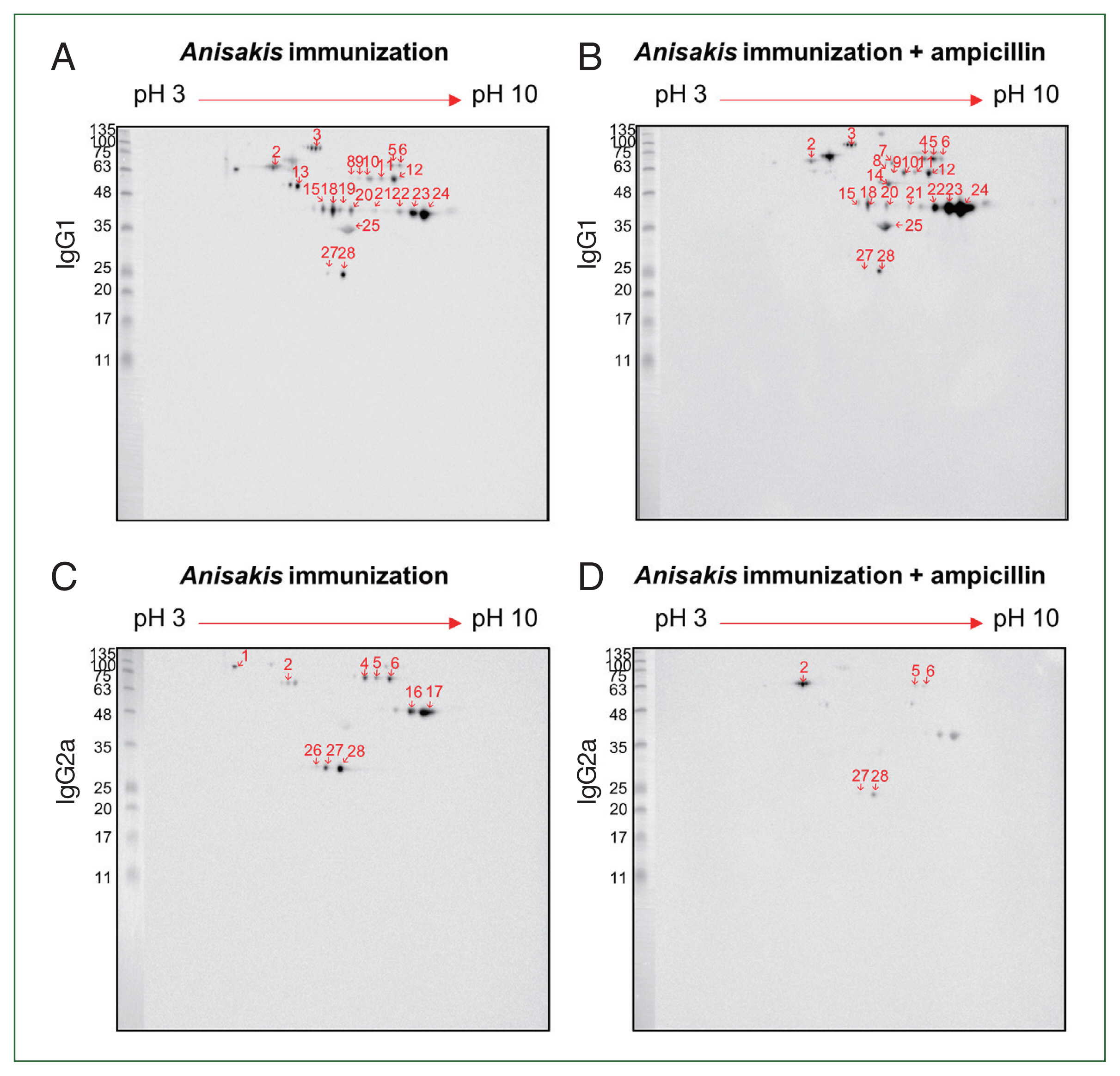
For improved resolution, we performed a 2-D immunoblot against
Anisakis extract with immunized mouse sera. Using
Anisakis-specific anti-mouse IgG1, spots 13 at 55 kDa and 19 at 46 kDa appeared only in the ampicillin-untreated
Anisakis-immunized group while spots 4 at 70 kDa and 7 at 70 kDa and 14 kDa appeared only in the ampicillin-treated
Anisakis-immunized group (
Fig. 5A, B). Using
Anisakis-specific anti-mouse IgG2a, spots 1, 16, 17, and 27 at 100, 48, 48, and 27 kDa, respectively, that were present in the ampicillin-untreated group disappeared in the ampicillin-treated group (
Fig. 5C, D). Furthermore, spots 5 and 6 at 70 kDa were reduced in the ampicillin-treated group. MS was performed to identify these proteins of selected spots in the 2-D immunoblot assay (
Table 1); spot 13 was identified at HSP70 (AIU38247.1) while spots 11 and 12 were identified as an unnamed protein product (VDK17787.1); these spots were detected in the 1-D western blotting MS analysis (
Table 3). These observations suggest that ampicillin treatment altered the immunized patterns of Th1- and Th2-inducing
Anisakis antigens.
Discussion
In this study, we observed that antibiotic treatment altered the mouse microbiome with subsequent changes to Ig production patterns following A. pegreffii immunization. Antibiotic treatment reduced the number of commensal microbiota species and diversity in the mouse microbiome and significantly decreased the abundance of Bacteroides. Furthermore, ampicillin treatment altered the immunized antigen patterns, with numerous IgG1 spots but fewer IgG2a spots observed in the ampicillin group than in the untreated groups.
We hypothesize that gut microbiome dysbiosis causes a Th1/Th2 imbalance, which further alters the antigen-immunization pattern. Supporting this, previous studies suggest that antibiotic administration can cause the reorganization of bacterial flora, inhibiting signaling through the Toll-like receptor 4 (TLR4) pathway [
32]. The inability to signal through TLR4 has been shown to contribute to the allergic immune reaction by markedly increasing the Th2 cytokine response to peanut-specific IgE [
32]. Consistent with this finding, we observed that an additional band related to Th2-related IgG1 was detected using western blotting while several spots were increased in the 2-D immunoblot.
Interestingly, changes in the microbiome caused by
Anisakis-immunization in mice without ampicillin treatment were also confirmed in the PCoA result. Samples from mice not treated with ampicillin after antigen injection exhibited a unique clustering pattern distinct from PBS-injected mice along the PCoA2 axis, hinting at potential sensitization-induced shifts in the gut microbiota composition. In a previous study, oral sensitization using OVA without any adjuvant produced marked allergic reactions and notable changes in the gut microbiome with disturbance of the gut flora [
33]. Therefore, we propose that allergic inflammation itself can trigger changes in the microbiome.
In mice, systemic levels of IgG1 antibodies closely correlate with an overall Th2 immune response profile, whereas those of IgG2a antibodies indicate an overall Th1 profile [
34]. Th1-dependent IFN-γ induces the production of IgG2a, whereas the Th2 cytokine, IL-4, induces the expression of IgG1 [
34]. For
A. pegreffii-specific IgG1, an additional band was detected in the ampicillin-treated group in the immunoblot assay but a respective band was not detected for
A. pegreffii-specific IgG2a in the ampicillin-treated group. Bands were detected as an unnamed protein and HSP70. HSP70 is a major immunodominant antigen in many parasitic infections and plays a key role in host-parasite interactions. Allergens associated with the HSP70 family have been found in a heterogeneous range of sources. Among others, HSP70 is an inhalant allergen for house dust mites, storage mites, biting midges, black flies, and cockroaches [
35].
Similarly, in the 2-D immunoblot, numerous IgG1 spots but fewer IgG2a spots were observed in the ampicillin-treated group than in the untreated groups. Enolase is a major cross-reacting allergen in plants, fungi, and fish and has also been recognized as an allergen in cockroaches [
36]. Hemoglobin, a novel major allergen of
Anisakis, is a ubiquitous protein found in prokaryotes, fungi, plants, and animals [
37].
Anisakis hemoglobin has been recognized by serum IgE levels in >50% of prospectively studied patients with cutaneous features after
Anisakis parasitism [
37].
In this study, we used
A. pegreffii to immunize mice as this is the most prevalent species infecting humans and fish in Korea [
38]. Although only
A. simplex is currently registered as an allergen by the WHO, studies have suggested that
A. pegreffii can also induce allergies. In a study comparing the major allergens of
A. pegreffii and
A. simplex, epitopes and motifs were found to be conserved in both species for Ani s 1, 2, and 9 [
39]. The similarities in the amino acid sequences of these allergens suggest potential similar health risks to humans. In our previous research,
A. pegreffii induced allergic asthma in a mouse model [
12].
The study has some limitations. We did not examine how the restoration of the mouse microbiome may contribute to the recovery of the antigen-immunization pattern. We also used pooled samples as the amount of sera from 1 mouse was insufficient for protein analysis. Consequently, in future studies, we plan to use other animal models to obtain enough sera to perform all experiments using individual samples. In addition, despite collecting 150 Anisakis specimens from a single fish and molecularly identifying 15 as A. pegreffii, we proceeded with immunological experiments on the remaining 135 specimens under the assumption they were also A. pegreffii. This approach overlooks the possibility that a single fish could be infected by multiple Anisakis species.
In conclusion, we observed that ampicillin treatment altered the gut microbiome of mice, altering the immune response to antigens from A. pegreffii. Thus, changes in the gut microbiome after ampicillin treatment affected the immune response to A. pegreffii antigens in mice. The findings have implications for developing new treatment strategies or vaccines against parasitic allergies.
Notes
-
Author contributions
Data curation: Choi JH
Formal analysis: Oh S
Funding acquisition: Kim JY
Investigation: Kim M
Project administration: Yi Mh
Supervision: Yong TS
Writing – original draft: Kim M
Writing – review & editing: Kim JY
-
The authors declare no competing interests.
Acknowledgement
This research was supported by Bumsuk Academic Research Fund in 2022.
Fig. 1Microbiome analysis of the mouse cecum. (A) Number of operational taxonomic units (OTUs). (B) Shannon index. (C) Principal coordinate analysis depicting differences in the taxonomic compositions of bacterial communities; control (yellow); ampicillin-untreated Anisakis-immunized group (red) and ampicillin-treated Anisakis-immunized group (blue). N.S., not significant. **P<0.01.
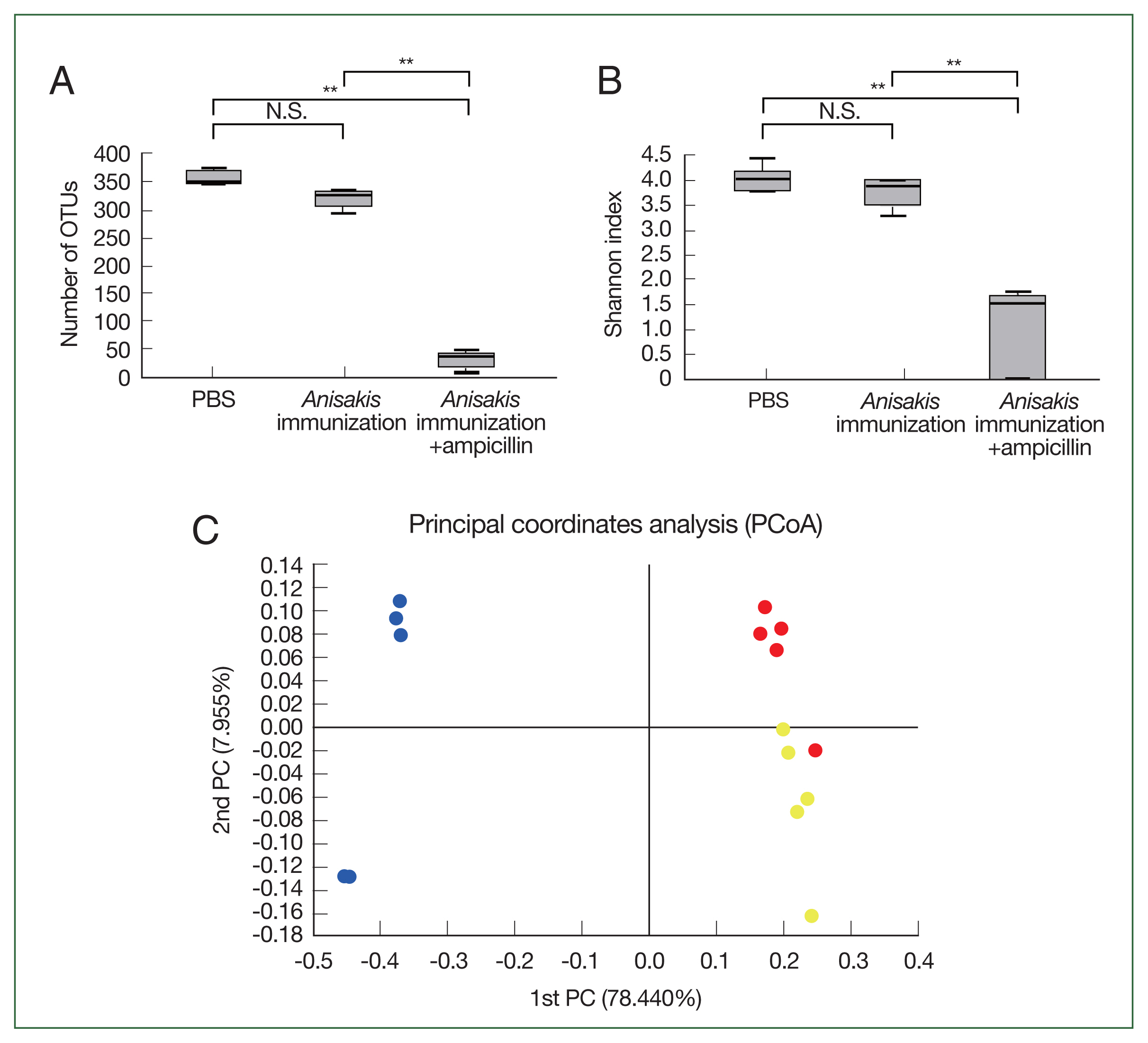
Fig. 2Relative abundance in the mouse cecum at the genus level.
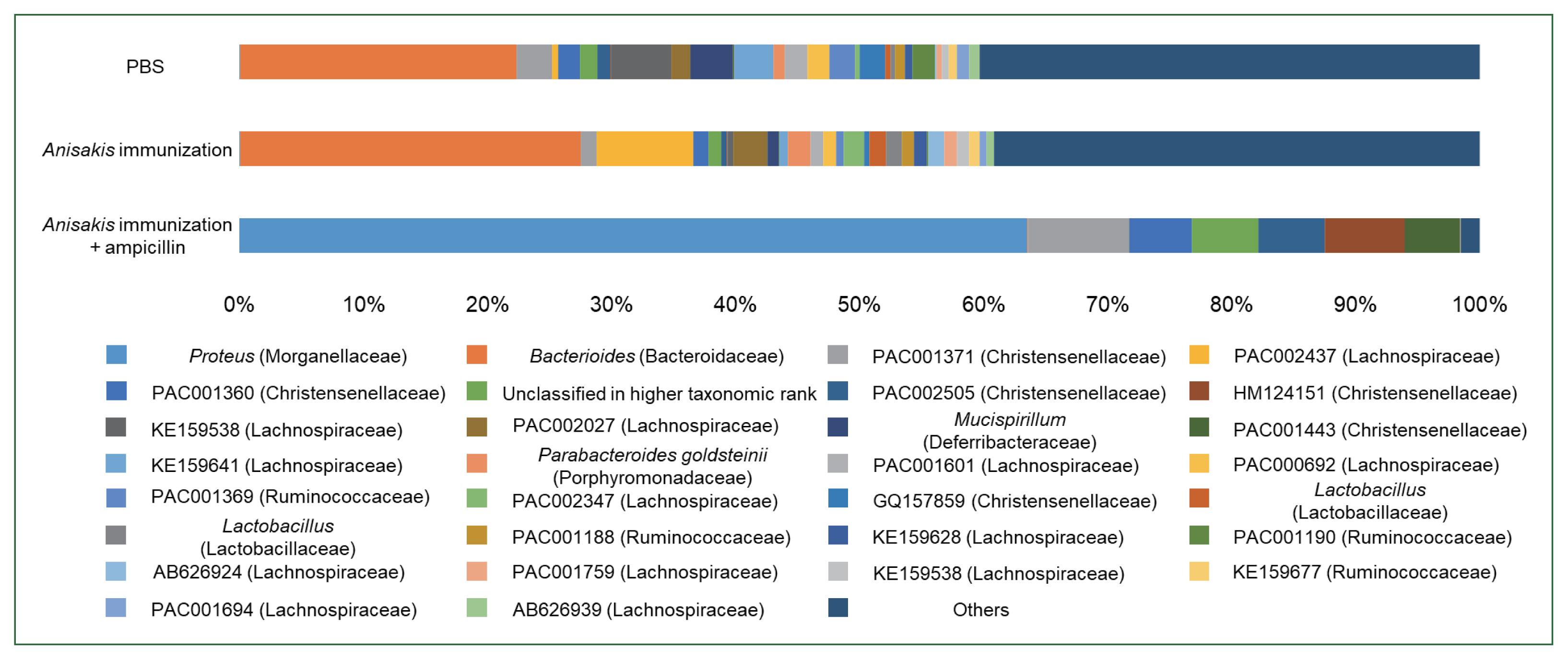
Fig. 3Analysis of Anisakis-specific antibody responses in mice sera samples using enzyme-linked immunosorbent assay (ELISA). (A) Anisakis-specific IgG1, (B) Anisakis-specific IgG2a. N.S., not significant. **P<0.01 and ***P<0.001.

Fig. 4Immunoblot analysis of Anisakis pegreffii protein extract with Anisakis-immunized mouse sera with or without ampicillin treatment. (A) Anisakis-specific IgG1 and (B) Anisakis-specific IgG2a.
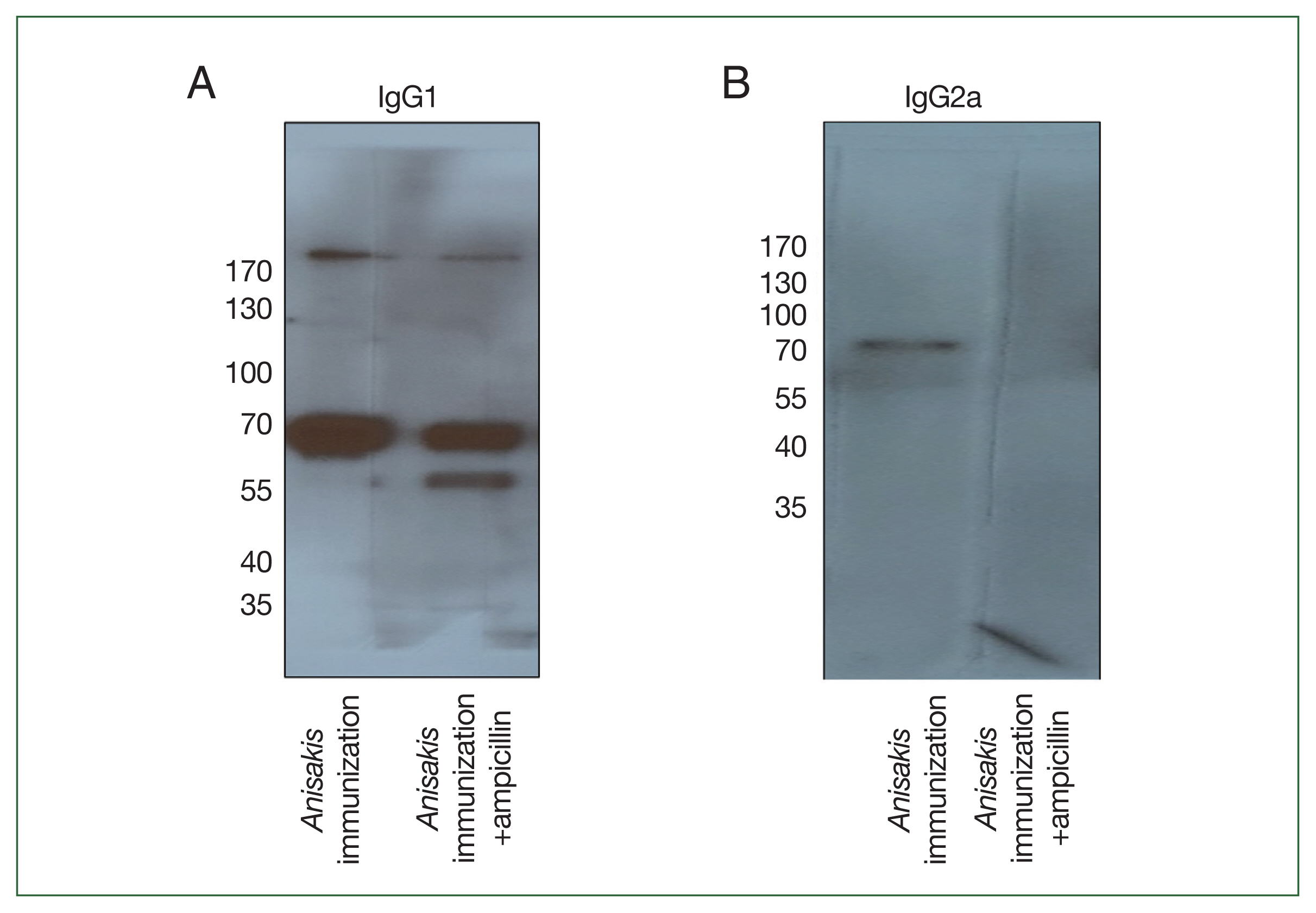
Fig. 5Two-dimensional immunoblot analysis of
Anisakis pegreffii extract with
Anisakis-immunized mouse sera.
Anisakis antigens reactive with IgG1 in
Anisakis-immunized mouse sera without/with ampicillin treatment (A and B, respectively).
Anisakis antigens reactive with IgG2a in
Anisakis-immunized mouse sera without/with ampicillin treatment (C and D, respectively). Red shows the identified number, which can be checked in
Table 1.
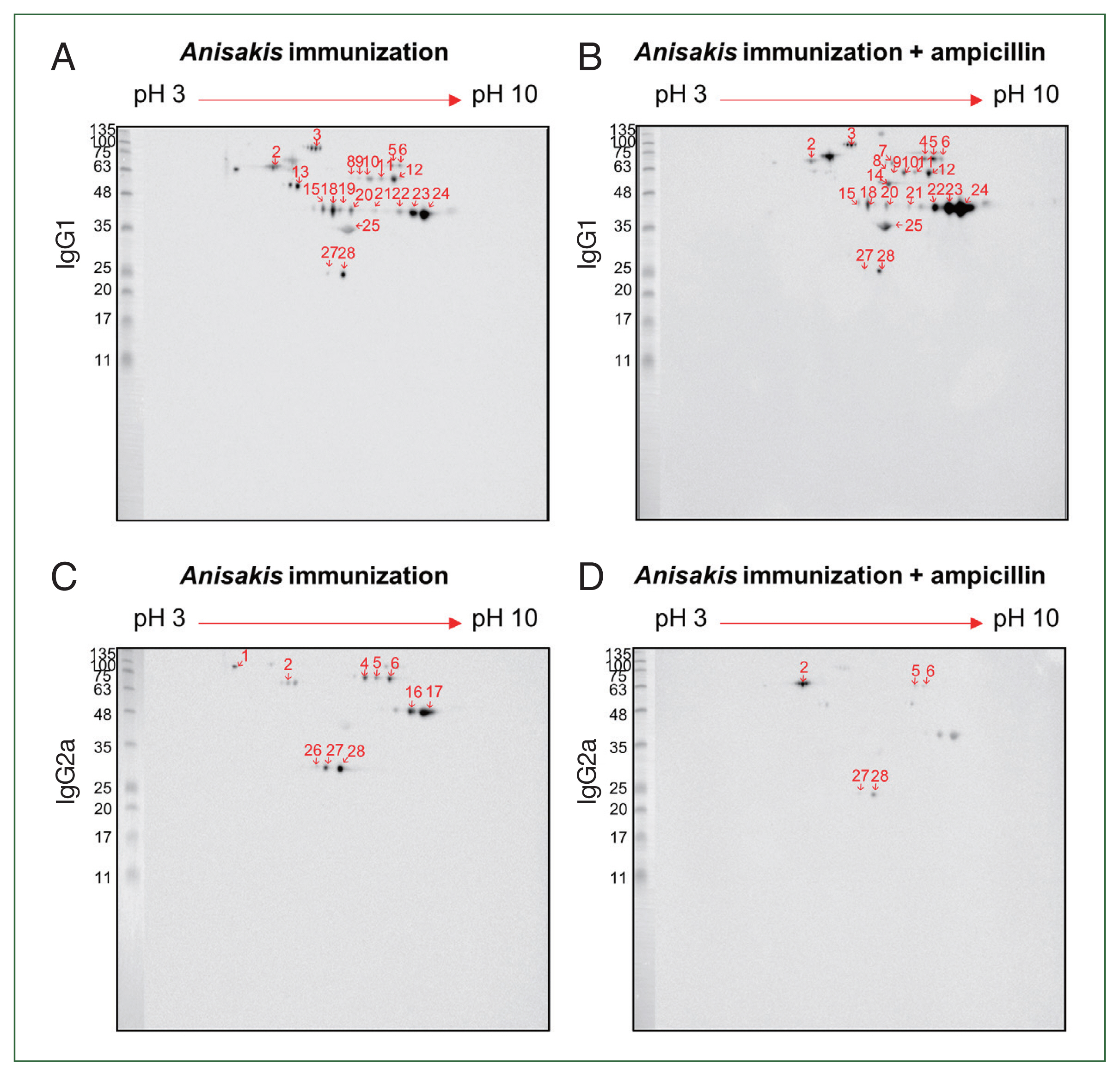
Table 1Allergen identification using LC-MS in 2D western blot analysis
Table 1
|
Spot no. |
NCBI BLAST |
Protein name |
Score |
Mass (m/z) |
|
1 |
P00761.1 |
RecName: Full=Trypsin; Flags: Precursor (Sus scrofa) |
54 |
25,078 |
|
2 |
VDK55667.1 |
78 kDa glucose-regulated protein (Anisakis simplex) |
345 |
75,647 |
|
3 |
2B5L_A |
Chain A, Crystal Structure of Ddb1 in Complex with Simian Virus 5 V Protein (Homo sapines) |
64 |
128,162 |
|
4 |
VDK50697.1 |
Chaperonin homolog Hsp-60, mitochondrial (Anisakis simplex) |
1,039 |
50,811 |
|
5 |
BAX39002.1 |
Hemoglobin (Anisakis pegreffii) |
197 |
39,305 |
|
6 |
|
No identification |
|
|
|
7 |
CAD43170.1 |
Enolase (Anisakis simplex) |
225 |
47,672 |
|
8 |
VDK47804.1 |
Unnamed protein product (Anisakis simplex) |
141 |
67,191 |
|
9 |
VDK54672.1 |
Unnamed protein product (Anisakis simplex) |
913 |
61,520 |
|
10 |
VDK54672.1 |
Unnamed protein product (Anisakis simplex) |
601 |
61,520 |
|
11 |
VDK17787.1 |
Unnamed protein product, partial (Anisakis simplex) |
61 |
32,612 |
|
12 |
VDK17787.1 |
Unnamed protein product, partial (Anisakis simplex) |
176 |
32,612 |
|
13 |
AIU38247.1 |
Heat shock protein 70 (Anisakis pegreffii) |
433 |
70,955 |
|
14 |
BAX39002.1 |
Hemoglobin (Anisakis pegreffii) |
58 |
39,305 |
|
15 |
VDK54672.1 |
Unnamed protein product (Anisakis simplex) |
687 |
61,520 |
|
16 |
VDK45319.1 |
Unnamed protein product (Anisakis simplex) |
213 |
53,638 |
|
17 |
VDK58822.1 |
Unnamed protein product (Anisakis simplex) |
61 |
52,879 |
|
18 |
VDK61442.1 |
Unnamed protein product (Anisakis simplex) |
72 |
8,957 |
|
19 |
|
No identification |
|
|
|
20 |
VDK58822.1 |
Unnamed protein product (Anisakis simplex) |
518 |
52,879 |
|
21 |
VDK58822.1 |
Unnamed protein product (Anisakis simplex) |
301 |
52,879 |
|
22 |
|
No identification |
|
|
|
23 |
CAD43170.1 |
Enolase (Anisakis simplex) |
240 |
47,672 |
|
24 |
CAD43170.1 |
Enolase (Anisakis simplex) |
634 |
47,672 |
|
25 |
CAD43170.1 |
Enolase (Anisakis simplex) |
885 |
47,672 |
|
26 |
CAD43170.1 |
Enolase (Anisakis simplex) |
1,758 |
47,672 |
|
27 |
Q0PGG4.1 |
RecName: Full=Actin, cytoplasmic 1; AltName: Full=Beta-actin;
Contains: RecName: Full=Actin, cytoplasmic 1, N-terminally processed (Bos grunniens) |
60 |
42,064 |
|
28 |
BAX39002.1 |
Hemoglobin (Anisakis pergeffii) |
181 |
39,305 |
Table 2Linear discriminant analysis effect size (LEfSe) analysis of differentially abundant bacterial taxa between control group, Anisakis immunization without ampicillin group and Anisakis immunization with ampicillin group. Only taxa meeting an LDA significant threshold of >4.5 are shown
Table 2
|
Taxon name |
Taxon rank |
LDA effect size |
P-value |
Relative abundance in control (%) |
Relative abundance in Anisakis immunization without ampicillin (%) |
Relative abundance in Anisakis immunization with ampicillin (%) |
|
Proteus vulgaris
|
Species |
5.54 |
0.00489 |
0.04 |
0.02 |
63.12 |
|
Bacteroides_uc |
Species |
5.16 |
0.00650 |
21.92 |
28.78 |
0.08 |
|
Protues
|
Genus |
5.17 |
0.00688 |
22.48 |
29.08 |
0.10 |
|
Bacteroides
|
Genus |
5.54 |
0.00489 |
0.04 |
0.02 |
63.12 |
|
Morganellaceae |
Family |
5.54 |
0.00489 |
0.04 |
0.02 |
63.14 |
|
Lachnospiraceae |
Family |
5.33 |
0.00888 |
43.70 |
43.90 |
0.10 |
|
Bacteroidaceae |
Family |
5.17 |
0.00688 |
22.48 |
29.08 |
0.10 |
|
Ruminococcaceae |
Family |
4.83 |
0.00628 |
14.16 |
12.62 |
0 |
|
Gammaproteobacteria |
Class |
5.54 |
0.00489 |
0.04 |
0.02 |
63.14 |
|
Bacteroidia |
Class |
5.20 |
0.00688 |
23.34 |
31.26 |
0.10 |
|
Enterobacterales |
Order |
5.54 |
0.00489 |
0.04 |
0.02 |
63.14 |
|
Bacteroidales |
Order |
5.20 |
0.00688 |
23.34 |
31.26 |
0.10 |
Table 3Allergen identification using MS analysis in 1D western blot analysis
Table 3
|
Band size |
Unused |
Total |
% Coverage |
Accession no. |
Name |
Peptides (95%) |
|
56 kDa |
41.01 |
41.01 |
69.3 |
VDK17787.1 |
Unnamed protein product |
22 |
|
28.36 |
28.36 |
37.1 |
VDK17834.1 |
Unnamed protein product |
15 |
|
|
70 kDa |
69.05 |
69.05 |
63.7 |
VDK17973.1 |
Unnamed protein product |
48 |
|
64.53 |
64.53 |
70.1 |
AIU38247.1 |
Heat shock protein 70 |
46 |
References
- 1. Sohn WM, Chai JY. Anisakiosis (Anisakidosis). In Palmer SR, Soulsby L, Torgerson PR, Brown WG, editors. Oxford Textbook of Zoonoses-Biology, Clinical Practice, and Public Health Control. 2nd ed. Oxford University Press; Oxford, UK. 2011. p. 774-786.
- 2. Bao M, Pierce GJ, Pascual S, González-Muñoz M, Mattiucci S, et al. Assessing the risk of an emerging zoonosis of worldwide concern: anisakiasis. Sci Rep 2017;7(1):43699.
https://doi.org/10.1038/srep43699
- 3. Kim JY, Yi MH, Yong TS. Allergen-like molecules from parasites. Curr Protein Pept Sci 2020;21(2):186-202.
https://doi.org/10.2174/1389203720666190708154300
- 4. Purello-D’Ambrosio F, Pastorello E, Gangemi S, Lombardo G, Ricciardi L, et al. Incidence of sensitivity to Anisakis simplex in a risk population of fishermen/fishmongers. Ann Allergy Asthma Immunol 2000;84(4):439-444.
https://doi.org/10.1016/S1081-1206(10)62278-8
- 5. Pravettoni V, Primavesi L, Piantanida M.
Anisakis simplex: current knowledge. Eur Ann Allergy Clin Immunol 2012;44(4):150-156.
- 6. Ciabattini A, Olivieri R, Lazzeri E, Medaglini D. Role of the microbiota in the modulation of vaccine immune responses. Front Microbiol 2019;10:1305.
https://doi.org/10.3389/fmicb.2019.01305
- 7. Qian LJ, Kang SM, Xie JL, Huang L, Wen Q, et al. Early-life gut microbial colonization shapes Th1/Th2 balance in asthma model in BALB/c mice. BMC Microbiol 2017;17(1):135.
https://doi.org/10.1186/s12866-017-1044-0
- 8. Watts AM, West NP, Zhang P, Smith PK, Cripps AW, et al. The gut microbiome of adults with allergic rhinitis is characterised by reduced diversity and an altered abundance of key microbial taxa compared to controls. Int Arch Allergy Immunol 2021;182(2):94-105.
https://doi.org/10.1159/000510536
- 9. Duong QA, Pittet LF, Curtis N, Zimmermann P. Antibiotic exposure and adverse long-term health outcomes in children: a systematic review and meta-analysis. J Infect 2022;85(3):213-300.
https://doi.org/10.1016/j.jinf.2022.01.005
- 10. Lee N, Kim WU. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med 2017;49(5):e340.
https://doi.org/10.1038/emm.2017.36
- 11. Baird FJ, Su X, Aibinu I, Nolan M, Sugiyama H. The Anisakis transcriptome provides a resource for fundamental and applied studies on allergy-causing parasites. PLoS Negl Trop Dis 2016;10(7):e0004845.
https://doi.org/10.1371/journal.pntd.0004845
- 12. Choi JH, Kim JY, Yi MH, Kim M, Yong TS.
Anisakis pegreffii extract induces airway inflammation with airway remodeling in a murine model system. Biomed Res Int 2021;2021:2522305.
https://doi.org/10.1155/2021/2522305
- 13. Ceylani T, Jakubowska-Doğru E, Gurbanov R, Teker HT, Gozen AG. The effects of repeated antibiotic administration to juvenile BALB/c mice on the microbiota status and animal behavior at the adult age. Heliyon 2018;4(6):e00644.
https://doi.org/10.1016/j.heliyon.2018.e00644
- 14. Baeza ML, Conejero L, Higaki Y, Martín E, Pérez C, et al.
Anisakis simplex allergy: a murine model of anaphylaxis induced by parasitic proteins displays a mixed Th1/Th2 pattern. Clin Exp Immunol 2005;142(3):433-440.
https://doi.org/10.1111/j.1365-2249.2005.02952.x
- 15. Bahk YY, Kim SA, Kim JS, Euh HJ, Bai GH, et al. Antigens secreted from Mycobacterium tuberculosis: identification by proteomics approach and test for diagnostic marker. Proteomics 2004;4(11):3299-3307.
https://doi.org/10.1002/pmic.200400980
- 16. Gobom J, Nordhoff E, Mirgorodskaya E, Ekman R, Roepstorff P. Sample purification and preparation technique based on nano-scale reversed-phase columns for the sensitive analysis of complex peptide mixtures by matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 1999;34(2):105-116.
https://doi.org/10.1002/(SICI)1096-9888(199902)34:2<105::AID-JMS768>3.0.CO;2-4
- 17. Koenig T, Menze BH, Kirchner M, Monigatti F, Parker KC, et al. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J Proteome Res 2008;7(9):3708-3717.
https://doi.org/10.1021/pr700859x
- 18. Kim JY, Kim EM, Yi MH, Lee J, Lee S, et al. Chinese liver fluke Clonorchis sinensis infection changes the gut microbiome and increases probiotic Lactobacillus in mice. Parasitol Res 2019;118(2):693-699.
https://doi.org/10.1007/s00436-018-6179-x
- 19. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114-2120.
https://doi.org/10.1093/bioinformatics/btu170
- 20. Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 2012;13:31.
https://doi.org/10.1186/1471-2105-13-31
- 21. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75(23):7537-7541.
https://doi.org/10.1128/AEM.01541-09
- 22. Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, et al. Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 2017;67(5):1613-1617.
https://doi.org/10.1099/ijsem.0.001755
- 23. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990;215(3):403-410.
https://doi.org/10.1016/S0022-2836(05)80360-2
- 24. Myers EW, Miller W. Optimal alignments in linear space. Comput Appl Biosci 1988;4(1):11-17.
https://doi.org/10.1093/bioinformatics/4.1.11
- 25. Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010;26(19):2460-2461.
https://doi.org/10.1093/bioinformatics/btq461
- 26. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27(16):2194-2200.
https://doi.org/10.1093/bioinformatics/btr381
- 27. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012;28(23):3150-3152.
https://doi.org/10.1093/bioinformatics/bts565
- 28. Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948;27:379-423.
- 29. Gower JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika 1966;53(3–4):325-338.
https://doi.org/10.2307/2333639
- 30. Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005;71(12):8228-8235.
https://doi.org/10.1128/AEM.71.12.8228-8235.2005
- 31. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60.
https://doi.org/10.1186/gb-2011-12-6-r60
- 32. Lee KH, Song Y, Wu W, Yu K, Zhang G. The gut microbiota, environmental factors, and links to the development of food allergy. Clin Mol Allergy 2020;18:5.
https://doi.org/10.1186/s12948-020-00120-x
- 33. Zhou C, Chen LL, Lu RQ, Ma WW, Xiao R. Alteration of intestinal microbiota composition in oral sensitized C3H/HeJ Mice is associated with changes in dendritic cells and T Cells in mesenteric lymph nodes. Front Immunol 2021;12:631494.
https://doi.org/10.3389/fimmu.2021.631494
- 34. Firacative C, Gressler AE, Schubert K, Schulze B, Müller U, et al. Identification of T helper (Th) 1-and Th2-associated antigens of Cryptococcus neoformans in a murine model of pulmonary infection. Sci Rep 2018;8(1):2681.
https://doi.org/10.1038/s41598-018-21039-z
- 35. Kochanowski M, Różycki M, Dąbrowska J, Bełcik A, Karamon J, et al. Proteomic and bioinformatic investigations of heat-treated Anisakis simplex third-stage larvae. Biomolecules 2020;10(7):1066.
https://doi.org/10.3390/biom10071066
- 36. Chuang JG, Su SN, Chiang BL, Lee HJ, Chow LP. Proteome mining for novel IgE-binding proteins from the German cockroach (Blattella germanica) and allergen profiling of patients. Proteomics 2010;10(21):3854-3867.
https://doi.org/10.1002/pmic.201000348
- 37. González-Fernández J, Daschner A, Nieuwenhuizen NE, Lopata AL, Frutos CD, et al. Haemoglobin, a new major allergen of Anisakis simplex. Int J Parasitol 2015;45(6):399-407.
https://doi.org/10.1016/j.ijpara.2015.01.002
- 38. Song H, Jung BK, Cho J, Chang T, Huh S, et al. Molecular identification of Anisakis larvae extracted by gastrointestinal endoscopy from health check-up patients in Korea. Korean J Parasitol 2019;57(2):207-211.
https://doi.org/10.3347/kjp.2019.57.2.207
- 39. Quiazon KM, Zenke K, Yoshinaga T. Molecular characterization and comparison of four Anisakis allergens between Anisakis simplex sensu stricto and Anisakis pegreffii from Japan. Mol Biochem Parasitol 2013;190(1):23-26.
https://doi.org/10.1016/j.molbiopara.2013.05.006