Abstract
We compared the DNA sequences of the genus Metagonimus: M. yokogawai, M. takahashii, and M. miyatai. We obtained 28S D1 ribosomal DNA (rDNA) and mitochondrial cytochrome c oxidase subunit I (mtCOI) fragments from the adult worms by PCR, that were cloned and sequenced. Phylogenetic relationships inferred from the nucleotide sequences of the 28S D1 rDNA and mtCOI gene. M. takahashii and M. yokogawai are placed in the same clade supported by DNA sequence and phylogenic tree analysis in 28S D1 rDNA and mtCOI gene region. The above findings tell us that M. takahashii is closer to M. yokogawai than to M. miyatai genetically. This phylogenetic data also support the nomination of M. miyatai as a separate species.
-
Key words: Metagonimus, Heterophyidae, polymerase chain reaction, sequence analysis, DNA, classification
INTRODUCTION
Metagonimiasis is an endemic intestinal trematode infection in Korea with 0.3% egg positives in the general population, i.e. 130,000 infected people (
Ministry of Health and Social Welfare, and Korea Association of Health, 1997). Human infection with the three
Metagonimus (Digenea: Heterophyidae) species is due to eating of raw fishes (intermediate hosts), and is regarded as a parasitic disease of importance to public health in the riverside areas of the southern and eastern coasts of Korea (
Yu et al., 1994;
Chai et al., 2000b;
Lee et al., 2002). Three species of the genus
Metagonimus are known in Korea (
Chai et al., 1991):
M. yokogawai (
Katsurada, 1912),
M. takahashii (
Suzuki, 1930), and
M. miyatai (
Miyata, 1941,
1944;
Saito et al., 1984,
1997). These three species are differentiated by their morphology (
Chai et al., 1991,
1998,
2000a), as they have different fish species as hosts (
Rim et al., 1996), different geographical distributions (
Kim et al., 1987;
Chai et al., 1993;
Yu et al., 1994;
Lee et al., 2002). In addition, they have different polymerase chain reaction-based restriction fragment length polymorphism (PCR-RFLP) patterns and random amplified polymorphic DNA (RAPD) patterns (
Yu et al., 1997a,
1997b), and simple sequence repeat anchored polymerase chain reaction (SSR-PCR) patterns of genomic DNA of ribosomal RNA and mitochondrial cytochrome c oxidase 1 (mtCOI) gene (
Yang et al., 2000). However, few studies have been conducted on the DNA sequences of
Metagonimus spp. In the present study, we compared the 28S D1 rDNA and mtCOI gene sequences of the three species of
Metagonimus distributed in the Republic of Korea, to determine their molecular phylogenies.
MATERIALS AND METHODS
Genomic DNA purification from worms
The adult specimens of
M. takahashii,
M. miyatai and
M. yokogawai species were obtained 7 days post-infection from rats (Sprague-Dawley, 4 to 6-wk-old). Metacercariae of each species were collected from Korean freshwater fishes,
Carassius auratus,
Zacco platypus, and
Plecoglossus altivelis, respectively (
Saito et al., 1997;
Yu et al., 1997b). Two adult trematodes of family Heterophyidae,
Pygidiopsis summa and
Stellantchasmus falcatus obtained from experimentally infected animals were included and
S. falcatus was used as an outgroup to infer phylogeny. Adult worms were stored at -70℃ until used. DNA was extracted using the phenol/chloroform method and precipitated in ethanol, as reported by Sambrook and Russell (
2001).
PCR was conducted using a mixed solution of extracted DNA as a template (0.01 µg/µl), primer, and ExTaq enzyme (TaKaRa Ex Taq Kit, TAKARA Shuzo Co., LTD. Japan) in a GeneAmp PCR System 9600 (Perkin Elmer, USA). The PCR reaction cycle consisted of 40 cycles of denaturation at 95℃ for 20 seconds, annealing at 55℃ for 30 seconds and extension at 72℃ for 30 seconds followed by a final extension of 6 minutes. The forward primer (JB10, 5' GATTACCCGCTGAACTTAAGCATA 3') consisted of the conserved region from the 21st-45th base pairs of 28S rDNA sequence of mouse. The reverse primer (JB9, 5' GCTGCATTCACAAACACCCCGACTC 3') was obtained from the 278th-302nd base pairs of the same gene (
Hassounna et al., 1984;
Qu et al., 1988;
Bowles and McManus, 1994). The PCR of mtCOI were performed using the above described method, but with different primers. The forward primer was JB3 (5' TTTTTTGGGCATCCTGAGGTTTAT 3') (2575), and the reverse primer was JB4.5 (5' TAAAGAAAGAACATAATGAAAATG 3') (3021). The numbers in brackets refer to the position of the 5' end of the primer from the nucleotide sequences of
Fasciola hepatica (
Bowles and McManus, 1994). The PCR condition used was the same as that described above, except for that annealing was done at 48℃ for 30 seconds.
The PCR products amplified with the primer sets were purified by gel extraction (QIAEX DNA Gel extraction Kit, QIAGEN Co., Germany) and were subcloned into the EcoRV site of a pT7Blue T-vector Kit plus ligase (Novagen Co., USA), according to the protocol from the supplier. For transformation, NovaBlue competent cells were used as a host cell (Novagen Co., WI, USA). The recombinant plasmid was screened using isopropyl-β-thiogalactoside (IPTG) and 5-bromo-4 chloro-3-indolyl-β-D-galactoside (X-gal). The cloned fragments in the recombinant plasmids were digested with
BamHI and
HindIII enzyme and purified from agarose gel using a QIAprep spin plasmid kit (QIAGEN Co.). DNA sequencing was performed by dideoxy chain termination method (
Sanger et al., 1977) using a Sequenase kit (ABI prism dye terminator cycle sequencing core kit, Perkin Elmer) and an automated DNA sequencer (Applied Biosystems model 373A, Perkin Elmer) on both strands with T3 and T7 primers. At least three clones were sequenced per sample with additional clones sequenced as necessary to resolve ambiguous sites.
For the analysis of DNA sequences, NCBI (National Center for Biotechnology Information) databases were used for homology analysis (BLAST2). We also calculated the fractional GC content of nucleic acid sequences using an EMBOSS GEECEE program in Sanger Institute, Cambridge, U.K. (
http://analysis.molbiol.ox.ac.uk/pise_html/geecee.html). All obtained sequences were aligned automatically using CLUSTAL W program (version 1.82, CLUSTAL W WWW Service at the European Bioinformatics Institute,
http://www.ebi.ac.uk/clustalw) for multiple sequence alignments while alignment gaps were treated as missing data (
Higgins et al., 1994, European Bioinformatics Institute,
http://www.ebi.ac.uk/clustalw/). Sequence format was Pearson (Fasta) (
Pearson and Lipman, 1988); sequence type was nucleotide (nt). The sequences were submitted to GenBank data base to get the accession numbers.
Phylogenetic trees were generated by TREEVIEW tree drawing software for windows (version 1.6.6,
Page, 1996) from aligned nucleotide sequence by CLUSTAL W program. To observe the tree type, we used Neighbor-Joining (NJ) as distance method (
Saitou and Nei, 1987), phylogeny inference package (PHYLIP) as parsimony method (version 3.4.,
Felsenstein, 1989,
1993). Alignment gaps were treated as missing data (
Hendy and Penny, 1982,
Hillis et al., 1996). The correction for nucleotide distance was done by the Kimura's 2 parameter method (
Kimura, 1980). The phylogenetic trees were outgroup rooted using the 28S D1 rDNA and mtCOI gene nucleotide sequences of
S. falcatus, because it represented a sister taxon.
RESULTS
The length of the 28S D1 region rDNA sequence of the three
Metagonimus species was 248 bp (adjusted for missing data) and its G+C content was 52% (
M. takahashii,
M. yokogawai and
M. miyatai). Nucleotide sequence differences among
Metagonimus species were less than 0.8% (2/248 bp), and that between the species, 0.4% (1/248 bp). There was no sequence gap in 28S D1 rDNA region (
Fig. 1). The length of the mtCOI sequence averaged 400 bp (398-403 bp, adjusted for missing data) with a G+C content ranged from 44% (
M. miyatai), 46% (
M. takahashii) to 47% (
M. yokogawai) (data not shown). Nucleotide sequence differences between species were 23.0% (92/400 bp) between
M. miyatai and
M. takahashii, 16.2% (65/400 bp) between
M. miyatai and
M. yokogawai, 13.2% (53/400 bp) between
M. takahashii and
M. yokogawai adjusted for missing data (
Fig. 2). Nucleotide gaps sequence differences were 2.5% (10/400 bp) between
M. miyatai and
M. takahashii, 2.7% (11/400 bp) between
M. miyatai and
M. yokogawai, and 2.4% (11/400 bp) between
M. takahashii and
M. yokogawai (
Fig. 2). The aligned sequences of three
Metagonimus species showed high similarities with other comparative human intestinal trematodes (
P. summa and
S. falcatus) for 28S D1 rDNA (95.0%, 234/248 bp and 94%, 233/248 bp) and the mtCOI gene (68.5%: 274/400 bp and 78.0%: 312/400 bp) from nucleotide BLAST database program in NCBI (
Figs. 1,
2).
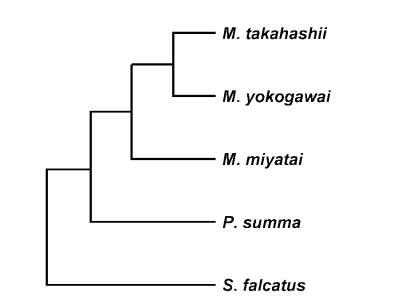
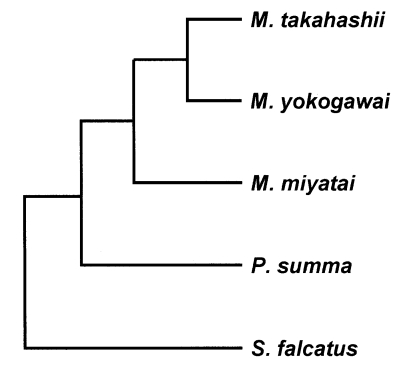
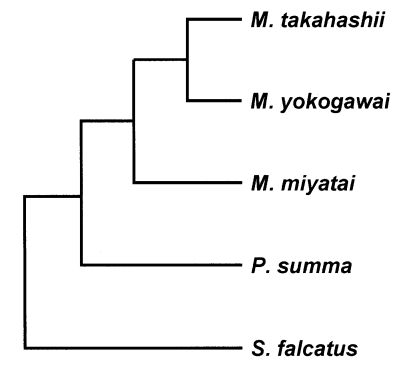
M. takahashii and
M. yokogawai placed in the same clade supported by DNA sequence and phylogenetic tree analysis for 28S D1 rDNA and mtCOI gene by neighbor-joining and parsimony method.
M. miyatai placed in different clade from two other
Metagonimus species (
Figs. 3,
4).
DISCUSSION
Our data coincided with the previously reported data, i.e., general morphology, PCR-RFLP, PCRRAPD and SSR-PCR patterns of the three
Metagonimus species. Rim et al. (
1996) reported that the general morphology of the three species of
Metagonimus was distinct and
M. miyatai type was distinctive from other species of
Metagonimus. Yu et al (
1997a &
1997b) suggested that the
M. miyata type had different DNA sequences from
M. yakogawai by RAPD and RFLP patterns of ITS1 and mtCOI gene. Yang et al. (
2000) showed that the three species of
Metagonimus had different genotypes. Our data supported the suggestions of previous genetic studies. However, sequence data did not coincide with the results of the chromosome analyses of these three
Metagonimus species. Lee et al. (
1999) also suggested that the karyology of
Metagonimus species had closer relationship between
M. miyatai and
M. takahashii (both 2n = 18) rather than between
M. miyatai and
M. yokogawai (2n = 32). This kind of discrepancy may be seen between karyologic and sequencing studies. We add a genetic evidence on the distinctness of
M. miyatai from
M. yokogawai and
M. takahashii.
Notes
-
This study was supported by the Academic Research Fund of Ministry of Education, Republic of Korea, 1997-1998 (Grant No. BM 97-204).
-
Sequence data from this article have been deposited with the EMBL/GenBank Data Libraries under accession numbers (Metagonimus miyatai: AF095333, M. takahashii: AF095332, M. yokogawai: AF095331).
References
- 1. Bowles J, McManus DP. Genetic characterization of the Asian Taenia, a newly described Taenid cestode of humans. Am J Trop Med Hyg 1994;50:33-44.
- 2. Chai JY, Guk SM, Han ET, et al. Surface ultrastructure of Metagonimus takahashii metacercariae and adults. Korean J Parasitol 2000a;38:9-15.
- 3. Chai JY, Han ET, Park YK, Guk SM, Kim JL, Lee SH. High endemicity of Metagonimus yokogawai infection among residents of Samchok-shi, Kangwon-do. Korean J Parasitol 2000b;38:33-36.
- 4. Chai JY, Huh S, Yu JR, Kook JN, Jung KC. An epidemiological study of metagonimiasis along the upperreaches of the Namhan River. Korean J Parasitol 1993;31:99-108.
- 5. Chai JY, Kang YJ, Choi SY, Guk SM, Yu JR, Lee SH. Surface ultrastructure of Metagonimus miyatai metacercariae and adults. Korean J Parasitol 1998;36:217-225.
- 6. Chai JY, Sohn WM, Kim MH, Hong ST, Lee SH. Three morphological types of the genus Metagonimus encysted in the dace, Tribolodon taczanowsjii, caught from the Sumjin river. Korean J Parasitol 1991;29:217-225.
- 7. Felsenstein J. PHYLIP-Phylogeny inference package (version 3.2). Cladistics 1989;5:164-166.
- 8. Felsenstein J. PHYLIP (Phylogeny Inference Package), version 3.5c. 1993. Seattle. Department of Genetics, University of Washington.
- 9. Hassounna N, Michot B, Bachellerie JP. The complete nucleotide sequence of mouse 28S rRNA gene. Implications for the process of size increase of the large subunit rRNA in higher eukaryotes. Nucleic Acids Res 1984;12:3563-3583.
- 10. Hendy MD, Penny D. Branch and bound algorithms to determine minimal evolutionary trees. Math Biosc 1982;59:277-290.
- 11. Higgins D, Thompson J, Gibson T, Thompson JD, Higgins DG, Gibson TJ. CLUSTA W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673-4680.
- 12. Hillis DM, Moritz C, Mable BK. In: Swofford Davis L., Olsen Gary J., Waddell Peter J., Hillis Davis M., editors. Chapter 11, Phylogenetic Inference. Molecular Systematics. 1996. 2nd ed. Sunderland, Massachusetts, USA. Sinauer Associates, Inc. Publishers; p. 407-514.
- 13. Katsurada F. Heterophyes in Japan, II. Creation of a new genus Metagonimus. Okayama Igakkai Zasshi 1912;273:768-778. (in Japanese).
- 14. Kim CH, Kim NM, Lee CH, Park JS. Studies on the Metagonimus fluke in the Daechung reservoir and the upper stream of Geum river. Korean J Parasitol 1987;25:69-82.
- 15. Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 1980;16:111-120.
- 16. Lee GS, Cho IS, Lee YH, et al. Epidemiological study of clonorchiasis and metagonimiasis along the Geumgang (river) in Okcheon-gun (County), Korea. Korean J Parasitol 2002;40:9-16.
- 17. Lee SU, Huh S, Park GM, Chai JY. A cytogenetic study of human intestinal parasites of the Genus Metagonimus (Digenea: Heterophyidae) in Korea. Korean J Parasitol 1999;37:237-241.
- 18. Ministry of Health and Social Welfare. Korea Association of Health. Prevalence of intestinal parasitic infections in Korea. The sixth report (Monographic series). 1997. Seoul, Korea. p. 1-71.
- 19. Miyata I. Supplementary discussion on the classification of the genus Metagonimus. Shokubutsu Oyobi Cobutsu 1941;9:533-534. (in Japanese).
- 20. Miyata I. Some discussions on the classification of the genus Metagonimus in Japan. Dobutsugaku Zasshi 1944;56(1-3):6-19. (in Japanese).
- 21. Page RDM. TREEVIEW: an application to display phylogenetic trees on personal computer. Comput Appl Biosci 1996;12:357-358.
- 22. Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 1988;85:2444-2448.
- 23. Qu LH, Nioloso M, Bachellerie JP. Phylogenetic calibration of the 5' terminal domain of large rRNA achieved by determining twenty eukaryotic sequences. J Mol Evol 1988;28:113-124.
- 24. Rim HJ, Kim KH, Joo KH. Classification and host specificity of Metagonimus spp. from Korean freshwater fish. Korean J Parasitol 1996;34:7-14.
- 25. Saito S. Species differentiation of genus Metagonimus. Reports on Meeting for Parasite Taxonomy and Morphology, Japan 1984;2:1-4.
- 26. Saito S, Chai JY, Kim KH, Lee SH, Rim HJ. Metagonimus miyatai sp. nov. (Digenea: Heterophyidae), a new intestinal trematode transmitted by fresh water fishes in Japan and Korea. Korean J Parasitol 1997;35:223-232.
- 27. Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 1987;4:406-425.
- 28. Sambrook J, Russell DW. Chapter 6. Molecular cloning: a laboratory manual. 2001. 3rd ed. New York, USA. Cold Spring Harbor Laboratory Press; p. 6.1-6.30.
- 29. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 1977;74:5463-5467.
- 30. Suzuki M. On Metagonimus yakogawai. Collected papers on studies of special animals in Okayama Prefecture. 1930. p. 146-168. p. XXII-XXIII Pls.(in Japanese).
- 31. Yang HJ, Guk SM, Han ET, Chai JY. Molecular differentiation of three species of Metagonimus by simple sequence repeat anchored polymerase chain reaction (SSR-PCR) amplification. J Parasitol 2000;86:1170-1172.
- 32. Yu JR, Chung JS, Chai JY. Differential RAPD patterns between Metagonimus yokogawai and Metagonimus Miyata type. Korean J Parasitol 1997a;35:295-298.
- 33. Yu JR, Chung JS, Huh S, Lee SH, Chai JY. PCR-RFLP pattern of three kinds of Metagonimus in Korea. Korean J Parasitol 1997b;35:271-276.
- 34. Yu JR, Kwon SO, Lee SH. Clonorchiasis and metagonimiasis in the inhabitants along Talchongang (River), Chungwon-gun. Korean J Parasitol 1994;32:267-269.
Fig. 1Sequences alignment of 28S D1 rDNA region of Metagonimus species. An asterisk (*) denotes an identical nucleotide position, and alignment gaps are indicated by a hyphen. Sequences for each species have been deposited in the GenBank databases (GenBank accession number: M. miyatai: AF095333, M. takahashii: AF095332, M. yokogawai: AF095331, P. summa: AF181885, S. falcatus: AF181886).

Fig. 2Sequences alignment of mtCOI region of Metagonimus species. An asterisk (*) denotes an identical nucleotide position, and alignment gaps are indicated by a hyphen. Sequences for each species have been deposited in the GenBank databases (GenBank accession number: M. miyatai: AF096232, M. takahashii: AF096231, M. yokogawai: AF096230, P. summa: AF181884, S. falcatus: AF181887).

Fig. 3Phylogenetic trees of Metagonimus species inferred from the 28S rDNA D1 gene nucleotide sequences by TREEVIEW program with the neighbor-joining and parsimony methods. The phylogenetic trees were outgroup rooted using the 28S D1 gene nucleotide sequence of Stellantchasmus falcatus.
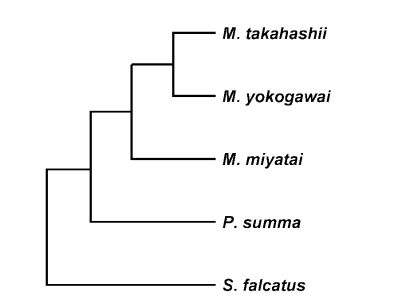
Fig. 4Phylogenetic trees of Metagonimus species inferred from the mtCOI gene nucleotide sequences by TREEVIEW program with the neighbor-joining and parsimony methods. The phylogenetic trees were outgroup rooted using the mtCOI gene nucleotide sequence of Stellantchasmus falcatus.
Citations
Citations to this article as recorded by

- Genetic Connectivity of Profilicollis altmani in Seabirds: Effects of Host Trophic Ecology on Chilean Coasts
Mauren Vergara, Sara M. Rodríguez, Maribet Gamboa
Austral Ecology.2026;[Epub] CrossRef - Molecular evidence for human Metagonimus kogai and M. saitoi infection in Korea: Detection of COI genes in the feces of riverside people along the Seomjin‑gang (river)
Eunsol Lee, Jong-Hun Choi, Yeong-Ju Lee, Seon-Ok Baek, Hee-Il Lee, Jung-Won Ju, Myeong-Ro Lee, Tae Yun Kim
Parasites, Hosts and Diseases.2026; 64(2): 123. CrossRef - Morphological and molecular data on helminths of Didelphis virginiana and Philander vossi (Mammalia: Didelphidae) from the Yucatán Peninsula, southeast Mexico
JESÚS ALONSO PANTI-MAY, ANYELA JACKELIN CHAN-CASANOVA, ELSY CANCHE-POOL, RAÚL TELLO-MARTÍN, HUGO RUIZ-PIÑA, HENRY CONCHA-GUILLERMO, OSCAR RETANA-GUIASCÓN, PEDRO PABLO MARTÍNEZ VEGA, JUAN CHABLÉ-SANTOS, ERENDIRA ESTRELLA-MARTÍNEZ, WILSON ISAIAS MOGUEL-CHIN
Zootaxa.2024; 5463(1): 1. CrossRef - Wood mouse (Apodemus sylvaticus L.) as intermediate host for Mesocestoides canislagopodis (Rudolphi, 1810) (Krabbe 1865) in Iceland
Damien Jouet, Aðalsteinn Örn Snæþórsson, Karl Skírnisson
Parasitology Research.2023; 122(9): 2119. CrossRef - Ancient DNA of Metagonimus yokogawai Recovered from Joseon Period Human Remains Newly Discovered at Goryeong County in South Korea
Chang Seok Oh, Jong Ha Hong, Jong Yil Chai, Mi Kyung Song, Ho-Jin Jang, Min Seo, Dong Hoon Shin
Acta Parasitologica.2022; 67(1): 539. CrossRef - Morphological and molecular identification of Cryptocotyle lingua metacercariae isolated from Atlantic cod (Gadus morhua) from Danish seas and whiting (Merlangius merlangus) from the English Channel
Maureen Duflot, Mélanie Gay, Graziella Midelet, Per Walter Kania, Kurt Buchmann
Parasitology Research.2021; 120(10): 3417. CrossRef - Metagonimus yokogawai Ancient DNA Recovered from 16th- to 17th-Century Korean Mummy Feces of the Joseon Dynasty
Jong Ha Hong, Min Seo, Chang Seok Oh, Jong-Yil Chai, Dong Hoon Shin
Journal of Parasitology.2020;[Epub] CrossRef - Characterization of the mitochondrial genome sequences of the liver fluke Amphimerus sp. (Trematoda: Opisthorchiidae) from Ecuador and phylogenetic implications
Jun Ma, Jun-Jun He, Cheng-Yan Zhou, Miao-Miao Sun, William Cevallos, Hiromu Sugiyama, Xing-Quan Zhu, Manuel Calvopiña
Acta Tropica.2019; 195: 90. CrossRef - Fishborne zoonotic heterophyid infections: An update
Jong-Yil Chai, Bong-Kwang Jung
Food and Waterborne Parasitology.2017; 8-9: 33. CrossRef - A molecular phylogeny of Asian species of the genus Metagonimus (Digenea)—small intestinal flukes—based on representative Japanese populations
Siritavee Pornruseetairatn, Hideto Kino, Takeshi Shimazu, Yukifumi Nawa, Tomáš Scholz, Jiraporn Ruangsittichai, Naowarat Tanomsing Saralamba, Urusa Thaenkham
Parasitology Research.2016; 115(3): 1123. CrossRef - Occurrence of Mesocestoides canislagopodis (Rudolphi, 1810) (Krabbe, 1865) in mammals and birds in Iceland and its molecular discrimination within the Mesocestoides species complex
Karl Skirnisson, Damien Jouet, Hubert Ferté, Ólafur K. Nielsen
Parasitology Research.2016; 115(7): 2597. CrossRef - Epidemiological and molecular data on heterophyid trematode metacercariae found in the muscle of grey mullets (Osteichthyes: Mugilidae) from Sardinia (western Mediterranean Sea)
Simonetta Masala, Maria Cristina Piras, Daria Sanna, Jong-Yil Chai, Bong-Kwang Jung, Woon-Mok Sohn, Giovanni Garippa, Paolo Merella
Parasitology Research.2016; 115(9): 3409. CrossRef - Advances in molecular diagnosis of parasitic enteropathogens
Shane Byrne, Jennifer M.B. Robson
Pathology.2015; 47(3): 234. CrossRef - Molecular characterization of Stictodora tridactyla (Trematoda: Heterophyidae) from Kuwait Bay using rDNA ITS and mtCO1
Wafa Y. Al-Kandari, Majed A. Alnaqeeb, Asha M. Isaac, Suzanne A. Al-Bustan
Parasitology Research.2015; 114(11): 4259. CrossRef - Molecular phylogeny of trematodes in Family Heterophyidae based on mitochondrial cytochrome c oxidase subunit I (mCOI)
Thapana Chontananarth, Chalobol Wongsawad, Siriwadee Chomdej, Duangduen Krailas, Jong Yil Chai
Asian Pacific Journal of Tropical Medicine.2014; 7(6): 446. CrossRef - Development of a polymerase chain reaction applicable to rapid and sensitive detection ofClonorchis sinensiseggs in human stool samples
Pyo Yun Cho, Byoung-Kuk Na, Kyung Mi Choi, Jin Su Kim, Shin-Hyeong Cho, Won-Ja Lee, Sung-Bin Lim, Seok Ho Cha, Yun-Kyu Park, Jhang Ho Pak, Hyeong-Woo Lee, Sung-Jong Hong, Tong-Soo Kim
Pathogens and Global Health.2013; 107(5): 253. CrossRef - Life cycle of Renylaima capensis, a brachylaimid trematode of shrews and slugs in South Africa: two-host and three-host transmission modalities suggested by epizootiology and DNA sequencing
Wilhelm F Sirgel, Patricio Artigas, M Dolores Bargues, Santiago Mas-Coma
Parasites & Vectors.2012;[Epub] CrossRef - To: Urusa Thaenkham and Yukifumi Nawa: Double Strand Problems: Reverse DNA Sequences Deposited in the DNA Database
Sun Huh
The Korean Journal of Parasitology.2010; 48(1): 91. CrossRef - Foodborne Intestinal Flukes in Southeast Asia
Jong-Yil Chai, Eun-Hee Shin, Soon-Hyung Lee, Han-Jong Rim
The Korean Journal of Parasitology.2009; 47(Suppl): S69. CrossRef - Molecular phylogeny of parasitic Platyhelminthes based on sequences of partial 28S rDNA D1 and mitochondrial cytochrome c oxidase subunit I
Soo-Ung Lee, Ha-Chung Chun, Sun Huh
The Korean Journal of Parasitology.2007; 45(3): 181. CrossRef - Development of Centrocestus armatus in different final hosts
Daisuke Kimura, Vachel Gay Paller, Shoji Uga
Veterinary Parasitology.2007; 146(3-4): 367. CrossRef - Fish-borne parasitic zoonoses: Status and issues
Jong-Yil Chai, K. Darwin Murrell, Alan J. Lymbery
International Journal for Parasitology.2005; 35(11-12): 1233. CrossRef