Abstract
Toxoplasma 3 main clonal lineages are designated as type I, II, and III; however, atypical and mixed genotypes were also reported. This study was conducted for detection of Toxoplasma gondii genotypes in rats (Rattus rattus) in Riyadh region, Saudi Arabia. PCR test on T. gondii B1 gene was conducted on ELISA IgM positive samples for confirmation of the infection. However, genetic analysis of the SAG2 locus was performed to determine T. gondii genotypes using PCR-RFLP technique. PCR test on T. gondii B1gene showed that 22 (81.5%) out of the 27 ELISA IgM positive samples have T. gondii DNA. Genotypic analysis shows that, of the total 22 PCR positive samples, only 13 (59.1%) were of type II, 7 (31.8%) were of type III, and 2 (9.1%) were of an unknown genotype. It is obvious that the prevalence of both type II and III is high in rats. No reports have been available on T. gondii genotypes among rats in Riyadh region, and only little is known about its seroprevalence in rats. Future studies on T. gondii genotypes in rats using multi-locus markers is needed in Riyadh region, Saudi Arabia for better understanding of T. gondii pathogenesis and treatment in humans and animals.
-
Key words: Toxoplasma gondii, genotypes, Rattus rattus, SAG2, PCR-RFLP, Saudi Arabia
INTRODUCTION
Toxoplasma gondii is an obligate intracellular parasite of worldwide distribution which infects almost all warm-blooded animals, including humans [
1]. It is estimated that nearly one-third of the human population in the world have infection with toxoplasmosis [
1]. Because cats are the natural definitive host, they play an important role in the epizootiology of toxoplasmosis. Rodents as generally considered as relevant markers to assess environmental contamination by toxoplasmosis and other pathogens and to estimate the risk of infection for definitive hosts [
2]. Behavioral studies comparing infected and uninfected mice have suggested that rodents lose their innate fear of cat odors when chronically infected with
T. gondii [
3], presumably enhancing the transmission of the parasite to its primary host.
In Saudi Arabia,
T. gondii seroprevalence in rats was 20% in Najran (Southwestern region) in
Meriones rex [
4], and in the Eastern region, 4 out of 5 rodents (mongooses) were found infected [
5]. However, in Riyadh, the
T. gondii seroprevalence was 12.5% (
Mus musculus) and 41.7% (
Rattus norvegicus), and the overall rate of infection was 35.6% [
6].
The population structure of
T. gondii includes 3 highly prevalent clonal lineages referred to as types I, II, and III, which differ greatly in virulence in the mouse model [
7]. Very little data regarding the
T. gondii genotypes is published in the Gulf and Saudi Arabia [
8]. Defining the
T. gondii genotypes has important implications for transmission, immunogenicity, and pathogenesis. The present study was conducted to detect
T. gondii genotypes in rats (
Rattus rattus) in Riyadh region, Saudi Arabia.
MATERIALS AND METHODS
Two hundred rats (
Rattus rattus) were collected from different regions in Riyadh. ELISA IgM assay was conducted for detection of
T. gondii positive samples followed by PCR amplification of the repetitive
T. gondii B1gene [
9]. Confirmed positive samples were analyzed using a nested-PCR that separately amplifies the 5'and 3'ends of
T. gondii SAG2 locus [
10,
11]. DNA was purified from blood using Puregene kit by following the instructions of the manufacturer (Gentra Systems, Minneapolis, Minnesota, USA). All procedures for animal experimentation used were approved by the Institutional Animal Ethics Committee, King Saud University, Riyadh, Saudi Arabia (KSU, NO.02/34/IAEC).
Briefly, PCR was performed using 10× PCR buffer (100 mM Tris-HCl; 500 mM KCl; 15 mM MgCl2; 0.01% gelatin) (Sigma Aldrich, St. Louis, Missouri, USA), 1 µM of each primer (Invitrogen-Life Technologies, Eggenstein, Germany), 2 U of Taq DNA polymerase (Invitrogen-Life Technologies) and 150-200 ng of DNA in a total reaction volume of 50 µl. PCR reactions were conducted in a GeneAmp® PCR System 9700 (Applied Biosystems, Foster City, California, USA). The first step of amplification was 10 min of denaturation at 95℃. This step was followed by 40 cycles, with 1 cycle consisting of 60 sec at 95℃, 45 sec at the annealing temperature for each pair of primers, and 60 sec at 72℃. The final cycle was followed by an extension step of 10 min at 72℃.
The 5'nd of the SAG2 locus was amplified using primers SAG2 F4 (5'CTACCTCGAACAGGAACAC3', SAG2 R4 (5'CATCAACAGTCTTCGTTGC3' at an annealing temperature of 61.5℃ for 55 sec. A second amplification was performed using 1 µl of the 1:10 diluted PCR products from the first amplification reaction as template for the second round, with similar PCR reaction mixture and conditions except for the primers, which were replaced by the internal set of primers, SAG2F (5'AAATGTTTCAGGTTGCTGC3', SAG2 R2 (5'CAAGAGCGAACTTGAACAC3'. Amplification of the 3'nd was performed with primers SAG2 F3 (5'CTGTTCTCCGAAGTGACTCC3', SAG2R3 (5'CAAAGCGTGCATTATCGC3' at an annealing temperature of 60℃ for 50 sec. For the second amplification, 1 µl of the 1:10 diluted PCR products were used as the template for the second amplification reaction, with internal primers SAG2F2 (5'TTCTCATGCCTCCGCTTC3', SAG2R (5'ACGTTTCACGAAGGCACAC3'. Similar PCR reaction mixture and conditions of the first amplification were used in the second round and 2% agarose gel was used to analyze the amplified PCR products. The amplified products were purified from primers, nucleotides, polymerases, and salts using, QlAquick PCR purification kit (QIAGEN Inc., Valencia, California, USA) according to the manufacturer's instructions.
Restriction digestion
Genotype analysis was performed using PCR-RFLP technique. The 5' end of the purified products of the SAG2 locus were digested using Sau3A1 endonuclease enzyme, whereas the 3' end purified products were digested using Hha1 endonuclease enzyme (Sigma-Aldrish). The products were analyzed using 2% agarose gel (Sigma-Aldrich). T. gondii strains RH (type I), LEG 96-1 (type II), and LEG-NJA (type III) were used as controls for lineage types. All the controls were generous gift from Dr. Darde ML, France.
RESULTS
Amplification of SAG2 locus
ELISA IgM assay showed that 27 samples (13.5%) out of 200 were positive. PCR test on
T. gondii B1 gene showed that, 22 (81.5%) out of the 27 ELISA IgM positive samples, have
T. gondii DNA (data not shown). The confirmed positive samples were subjected to SAG2 amplification. A nested-PCR method based on the polymorphic SAG2 locus, separately amplifying the 5'and 3'ends of the loci was used [
10,
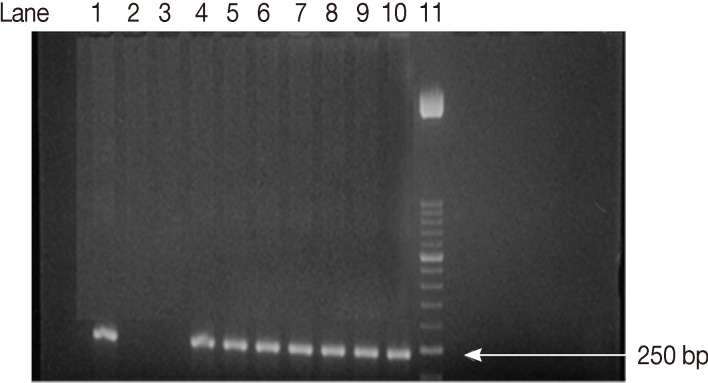
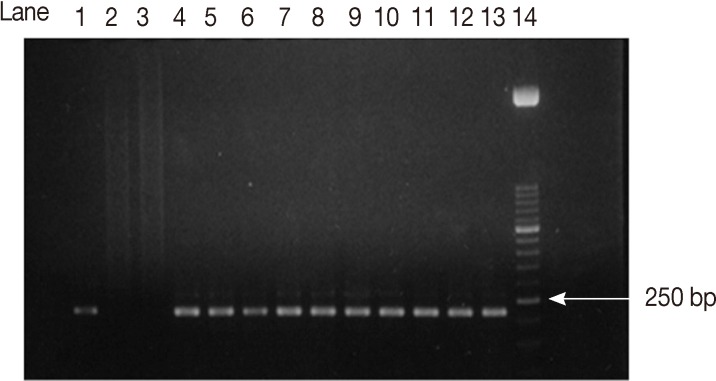
11]. The amplified products obtained 241 bp (
Fig. 1) and 221bp (
Fig. 2) fragments for the 5'and 3'ends of the loci, respectively.
The SAG2 PCR products were digested with restriction enzymes, and their type was determined according to the restriction patterns after separation by agarose gel electrophoresis. Three T. gondii strains served as lineage controls: RH (type I), LGE 96-1 (type II), and LEG-NJA (type III).
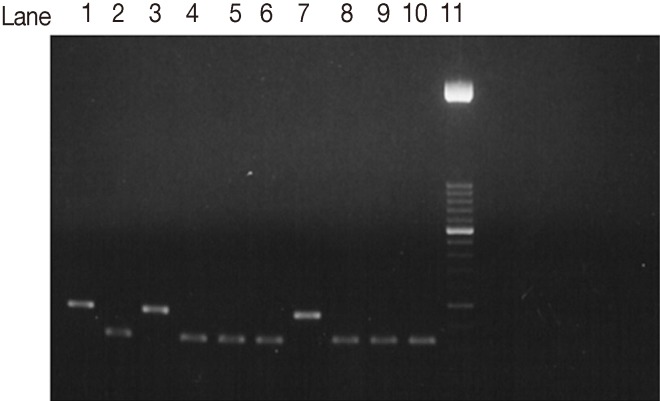
The SAG2 completely characterized samples were 17 out of 22 PCR positive samples (77.3%). When they were subjected to restriction digestion, 13 out of the 17 (76.5%) were characterized as genotype II (
Fig. 3) (3'end fragments were digested by
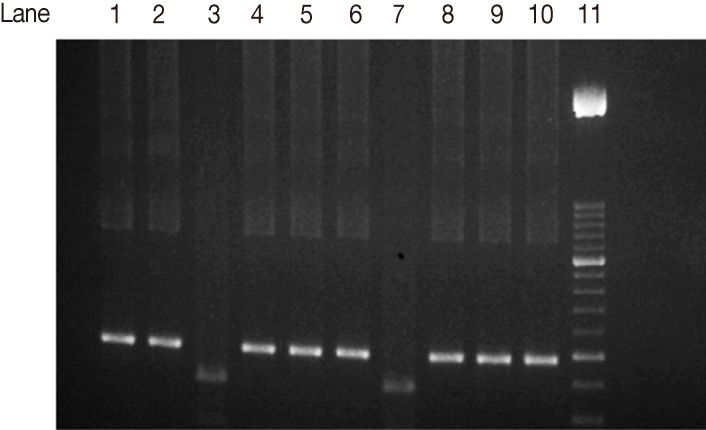
Hha1 endonuclease enzyme). Four out of 17 (23.5%) were characterized as type III (
Fig. 4) (5'end fragments were digested by
Sau3A1 enzyme.
The rest of the samples (5 samples) were partially characterized, as the 5' end only could be amplified. Of those fragments, 3 were Sau3A1 digested and characterized as type III, the other 2 samples were not digested and considered of unknown genotype.
Overall, of the total 22 PCR positive samples, 13 (59.1%) were type II, and 7 (31.8%) were type III, and 2 (9.1%) were of unknown genotype (
Table 1).
DISCUSSION
In the present study, PCR test on
T. gondii B1gene showed that 22 (81.5%) out of the 27 ELISA IgM positive samples had
T. gondii DNA. Only very little data were reported on the
T. gondii genotypes in the Middle East, and to our knowledge, no reports on the
T. gondii genotypes studies in rats as well. PCR-RFLP of SAG2 was used to classify
T. gondii strains. RFLP shows a good sensitivity level (5 parasites/sample) permitting direct typing on clinical samples [
12]. RFLP on SAG2 locus has been used for genotyping of
T. gondii isolates from various animals in Brazil [
13,
14], other parts of South America [
15], and Africa [
16].
In the present study, we found that most of the rats were infected with
T. gondii type II (59.1%; 13 out of 22 PCR positive samples) followed by type III (31.8%; 7 out of 22). To our knowledge, no genotyping studies were conducted in rats in Saudi Arabia. However, there is 1 published study concerning
T. gondii genotypes in Madinah, Saudi Arabia in humans and farm animals. The authors reported that, the type II isolate was the predominant one (45.1%), and the other genotypes I and III were detected in 20.9% and 21.9% of cases, respectively. They also found that, 12.1% of the samples were of unknown genotypes [
17]. In their study, most of the cases were harboring type II, while type I and III were almost equally distributed. The predominance of type II, followed by type III, in the present study agrees, to some extent, to the other from Madinah. However, we found higher percentages of type II (59.1%) and III (31.8%), compared to 45.1% and 21.9%, respectively, in the previous study in Madinah. In UAE and Qatar, type II was found in sand cats in addition to atypical strains [
18]. Worldwide,
T. gondii type II was reported in many food animals [
19]. However, strains from infected animals in Egypt [
16] and Iran [
20] were typed as type II and III, respectively. The high prevalence of type II in animals may underlie the prevalence of
T. gondii genotypes causing human disease.
None of the samples was harboring
T. gondii type I in the present study; however, it was detected in Madinah [
17] as mentioned previously, the discrepancy could be explained by that different species could harbor different
T. gondii genotypes, and also the source of infection for the subjects in each study could be different. Other explanation could be that the rat's habitats are different from those of humans and domestic animals, and the feeding habits and the route of infection could also be different. Generally, the lack of information on
T. gondii genotypes in Saudi Arabia specifically and the Middle East in general, makes it difficult to interpret the results of the present study. The absence of the type I strain in countries in the Eastern Mediterranean region in general was reported [
16]. We found that 2 (9.1%) samples were of unknown genotype, a finding which agrees to the other from Madinah, in which they found 12.1% were of unknown genotypes [
17].
The present study is the first report of T. gondii genotyping in wild rats in Saudi Arabia and Gulf region, the results provided preliminary data for further approaches in epidemiology and genotyping of T. gondii in these regions. It will be noteworthy to see whether eradication of wild rats in Saudi Arabia will affect the seroprevalence and genotype distribution of T. gondii, as rats are abundant in Saudi Arabia. Future studies using analysis of multiple T. gondii markers are necessary for new parasite typing identification in Saudi Arabia and the East mediterranean regions.
King Abdul-Aziz City for Science and Technology (KACST)10-ENV993-02
Notes
-
We have no conflict of interest related to this study.
ACKNOWLEDGMENT
This work was supported by the National Plan for Science and Technology (NPST) funded by King Abdul-Aziz City for Science and Technology (KACST) through project number 10-ENV993-02.
References
- 1. Dubey JP, Lindsay DS, Speer CA. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 1998;11:267-299.
- 2. Reperant LA, Hegglin D, Tanner I, Fischer C, Deplazes P. Rodents as shared indicators for zoonotic parasites of carnivores in urban environments. Parasitology 2009;136:329-337.
- 3. Vyas A. Parasite-augmented mate choice and reduction in innate fear in rats infected by Toxoplasma gondii. J Exp Biol 2013;216:120-126.
- 4. Morsy TA, El-Bahrawy AF, El-Dakhil MA. Ecto-and blood parasites affecting Meriones rex trapped in Najran, Saudi Arabia. J Egypt Soc Parasitol 2001;31:399.
- 5. Al Dakhil MA, Morsy TA. Natural Toxoplasma infection sought in the Indian grey mongoose (H. edwardsi Greffroy, 1818) trapped in the eastern region, Saudi Arabia. J Egypt Soc Parasitol 1996;26:645.
- 6. Morsy TA, Sabry AH, Habib KS, Arafa MA, El-Bahrawy AF, Al-Dakhil MM. Antibodies against Toxoplasma in commensal rodents. J Egypt Soc Parasitol 1994;24:279-284.
- 7. Sibley LD, Ajioka JW. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu Rev Microbiol 2008;62:329-351.
- 8. Wang L, Cheng HW, Huang KQ, Xu YH, Li YN, Du J, Yu L, Luo QL, Wei W, Jiang L, Shen JL. Toxoplasma gondii prevalence in food animals and rodents in different regions of China: isolation, genotyping and mouse pathogenicity. Parasit Vectors 2013;6:273.
- 9. Burg JL, Grover CM, Pouletty P, Boothroyd JC. Direct and sensitive detection of a pathogenic protozoan, Toxoplasma gondii, by polymerase chain reaction. J Clin Microbiol 1989;27:1787-1792.
- 10. Howe DK, Honoré S, Derouin F, Sibley LD. Determination of genotypes of Toxoplasma gondii strains isolated from patients with toxoplasmosis. J Clin Microbiol 1997;35:1411-1414.
- 11. Fuentes I, Rubio JM, Ramírez C, Alvar J. Genotypic Characterization of Toxoplasma gondii strains associated with human toxoplasmosis in Spain: direct analysis from clinical samples. J Clin Microbiol 2001;39:1566-1570.
- 12. Khan A, Su C, German M, Storch GA, Clifford DB, Sibley LD. Genotyping of Toxoplasma gondii strains from immunocompromised patients reveals high prevalence of type I strains. J Clin Microbiol 2005;43:5881-5887.
- 13. de A Dos Santos CB, de Carvalho AC, Ragozo AM, Soares RM, Amaku M, Yai LE, et al. First isolation and molecular characterization of Toxoplasma gondii from finishing pigs from São Paulo State, Brazil. Vet Parasitol 2005;131:207-211.
- 14. Dubey JP, Graham DH, Blackston CR, Lehmann T, Gennari SM, Ragozo AMA. Biological and genetic characterisation of Toxoplasma gondii isolates from chickens (Gallus domesticus) from São Paulo, Brazil: unexpected findings. Int J Parasitol 2002;32:99-105.
- 15. Dubey JP, Venturini MC, Venturini L, Piscopo M, Graham DH, Dahl E. Isolation and genotyping of Toxoplasma gondii from free-ranging chickens from Argentina. J Parasitol 2003;89:1063-1064.
- 16. Dubey JP, Graham DH, Dahl E, Hilali M, El-Ghaysh A, Streekumar C. Isolation and molecular characterization of Toxoplasma gondii from chickens and ducks from Egypt. Vet Parasitol 2003;114:89-95.
- 17. Abd El-Aal AA, Habib FA, Sheikh B, Harak M, Shalaby SA. Toxoplasma genotyping among infected human and animal hosts using PCR-restriction fragment length polymorphism: study in Al-Madinah, Saudi Arabia. Int J Health Sci 2010;III:2.
- 18. Dubey JP, Pas A, Rajendran C, Kwok OC, Ferreira LR, Martins J, Hebel C, Hammer S, Su C. Toxoplasmosis in sand cats (Felis margarita) and other animals in the breeding centre for endangered Arabian wildlife in the United Arab Emirates and Al Wabra wildlife preservation, the State of Qatar. Vet Parasitol 2010;20:195-203.
- 19. Mondragon R, Howe DK, Dubey JP, Sibley LD. Genotypic analysis of Toxoplasma gondii isolates in pigs. J Parasitol 1998;84:639-641.
- 20. Fazaeli A, Carter PE, Darde ML, Pennington TH. Molecular typing of Toxoplasma gondii strains by GRA6 gene sequence analysis. Int J Parasitol 2000;30:637-642.
Fig. 1Agarose gel electrophoresis analysis of SAG2 PCR amplification products. Second amplification of the 5'end of SAG2 locus. Lane 1, positive control. Lanes 2, 3, negative controls. Lanes 4-10, some positive samples (241 bp). Lane 11, 50-bp DNA marker.

Fig. 2Agarose gel electrophoresis analysis of SAG2 PCR amplification products. Second amplification of the 3'end of SAG2 locus. Lane 1, positive control. Lanes 2, 3, negative controls. Lanes 4-13, some positive samples (221 bp). Lane 14, 50-bp DNA marker.

Fig. 3Agarose gel electrophoresis analysis of SAG2 PCR amplification products and restriction digests from T. gondii-infected samples. Hha1 restriction digestion of 3'end amplification products of SAG2 locus. Lane 1, type I strain. Lane 2, Type II strain. Lane 3, Type III strain. Lanes 4-6, 8-10, samples of type II strains. Lane 7, non-type II sample. Lane 11, 50-bp DNA marker.

Fig. 4Agarose gel electrophoresis analysis of SAG2 PCR amplification products and restriction digests from T. gondii-infected samples. Sau3A1 restriction digestion of the 5'end amplification products of SAG2 locus. Lane 1, type I strain. Lane 2, type II strain. Lane 3, type III strain. Lanes 4-6, 8-10, samples of non-type III strains. Lane 7, type III sample. Lane 11, 50-bp DNA marker.

Table 1.Blood samples of rats and characterized genotypes of T. gondii strains
Table 1.
|
Number of blood sample |
PCR RFLP results
|
Toxoplasma type |
|
SAG2-3´ end |
SAG2-5´ end |
|
1 |
+ |
- |
II |
|
2 |
+ |
- |
II |
|
3 |
+ |
- |
II |
|
4 |
+ |
- |
II |
|
5 |
- |
+ |
III |
|
6 |
- |
+ |
III |
|
7 |
NA |
- |
Non type III |
|
8 |
+ |
- |
II |
|
9 |
NA |
+ |
III |
|
10 |
+ |
- |
II |
|
11 |
+ |
- |
II |
|
12 |
+ |
- |
II |
|
13 |
+ |
- |
II |
|
14 |
- |
+ |
III |
|
15 |
NA |
+ |
III |
|
16 |
NA |
- |
Non type III |
|
17 |
+ |
- |
II |
|
18 |
+ |
- |
II |
|
19 |
+ |
- |
II |
|
20 |
- |
+ |
III |
|
21 |
NA |
+ |
III |
|
22 |
+ |
- |
II |