Abstract
The genetic diversity of Schistosoma haematobium remains largely unstudied in comparison to that of Schistosoma mansoni. To characterize the extent of genetic diversity in S. haematobium among its definitive host (humans), we collected S. haematobium eggs from the urine of 73 infected schoolchildren at 5 primary schools in White Nile State, Sudan, and then performed a randomly amplified polymorphic DNA marker ITS2 by PCR-RFLP analysis. Among 73 S. haematobium egg-positive cases, 13 were selected based on the presence of the S. haematobium satellite markers A4 and B2 in their genomic DNA, and used for RFLP analysis. The 13 samples were subjected to an RFLP analysis of the S. haematobium ITS2 region; however, there was no variation in size among the fragments. Compared to the ITS2 sequences obtained for S. haematobium from Kenya, the nucleotide sequences of the ITS2 regions of S. haematobium from 4 areas in Sudan were consistent with those from Kenya (> 99%). In this study, we demonstrate for the first time that most of the S. haematobium population in Sudan consists of a pan-African S. haematobium genotype; however, we also report the discovery of Kenyan strain inflow into White Nile, Sudan.
-
Key words: Schistosoma haematobium, Sudan, ITS2, PCR-restriction fragment length polymorphism (PCR-RFLP)
INTRODUCTION
Schistosomiasis is a chronic and debilitating disease, second only to malaria in terms of parasite-induced human morbidity and mortality. Among the species of schistosomes infecting humans,
Schistosoma haematobium is responsible for the largest number of infections in sub-Saharan Africa; an estimated 112 million people are infected with this species, which is more than double the estimated figure for
Schistosoma mansoni [
1]. In Sudan,
S. haematobium infection is dominant, although both
S. haematobium and
S. mansoni are found [
2,
3].
Studies of the genetic diversity of natural
Schistosoma populations are complicated by the fact that adult worms are inaccessible in the cardiovascular system of the mammalian host. Molecular epidemiological studies of schistosomiasis have provided opportunities to investigate many important topics such as the contribution of parasite genetics to variation in disease burden and pathology, the genetic consequences of various control activities for parasite populations, patterns of recruitment and transmission in endemic areas, and the likely evolution and spread of drug resistance [
4,
5]. Previous reports have demonstrated that the second internal transcribed spacer (ITS2) of
S. haematobium sequences from Kenya were nearly identical (99%) to conspecific sequences from Egypt, Mali, and Niger [
6], and
Schistosoma japonicum in China was highly genetically diverse by cloning ITS1-ITS2 sequences according to the location [
7]. Recently, it was reported that a high level of genetic variability of
S. haematobium in the populations from Mali and Nigeria [
8].
However, the genetic diversity of
S. haematobium remains largely unstudied in comparison to
S. mansoni [
9], primarily because of the more demanding conditions for laboratory maintenance and lack of available molecular markers [
10]. Moreover, there were no reports about the extent of genetic diversity of
S. haematobium within its definitive host, humans in Sudan. Here, to characterize the genotype of
S. haematobium in an infected population and to identify potential associations with parasite diversity, we collected
S. haematobium eggs from 73 infected children at 5 schools in Sudan and performed genotyping using a polymorphic microsatellite marker, ITS2 region.
MATERIALS AND METHODS
Ethical statement
This study protocol was reviewed and approved by the institutional review board of the Korea Association of Health Promotion (acceptance no. 12-C-01) and was also approved by the National Control Program for Schistosomiasis and Soil-Transmitted Helminths, Federal Ministry of Health, Sudan. Before collecting the urine samples, informed verbal consent was obtained from each child with the presence of school teachers.
Study areas and collection of S. haematobium eggs
S. haematobium eggs were collected from 73 children at 5 primary schools in February of 2013. Five primary schools were selected in the Al Jabalain locality of White Nile, Sudan. All schools were located adjacent to the White Nile River, and they were located at Al Hidaeb, Al Zealet, Jazeera Aba, Khour Ajwal, and Al Sidding villages. From each child, 10-15 ml of urine specimen was collected, and transferred to the Schistosomiasis Control Center established by the Korea International Cooperation Agency (KOICA) in Kosti, White Nile State, Sudan. In the Center, urine samples were centrifuged at 1,500 rpm for 5 min, and the pellets were examined for eggs of S. haematobium by microscopy.
Preparation of S. mansoni genomic DNA
Adult S. mansoni specimens were obtained from Department of Environmental Medical Biology and Institute of Tropical Medicine, Yonsei University College of Medicine, Korea, and the fluke was stored at -70˚C. For DNA isolation, adult S. mansoni was cut into 20 mg tissue samples using a sterile scalpel. Genomic DNA was isolated from adult S. mansoni worms using a G-DEX™ genomic DNA extraction kit (iNtRON Biotechnology, Seoul, Korea) according to the manufacturer’s instructions.
Multiplex PCR assay
Genomic DNA was isolated from
S. haematobium eggs using a G-DEX™ genomic DNA extraction kit (iNtRON Biotechnology) according to the manufacturer’s instructions. PCR was performed with primers specific for 2 microsatellites markers of
S. haematobium, A4 and B2, as described previously [
7]. All amplifications were performed using TaKaRa Ex Taq DNA Polymerase (Takara Bio Inc., Otsu, Japan) with the following primers: A4(F) (5´-CGA ACT CCA ACG AGC ATC-3´), A4(R) (5´-GGG TGT GGG AAT GAC TTG-3´), B2(F) (5´-AAG CCG ACC ATT TGA CTC-3´), and B2(R) (5´-GTT GCT GTT GAT GAC GAT G-3´). The following conditions were carried out with an initial denaturation step of 94˚C for 30 sec, followed by 30 cycles of denaturation at 98˚C for 10 sec, annealing at 60˚C for 30 sec, and extension at 72˚C for 30 sec. The multiplex PCR products were then electrophoresed on a 2% agarose gel, stained with ethidium bromide, and visualized under UV light.
PCR amplification of the ITS2 fragment (ITS2-PCR) from S. haematobium eggs was performed in a 100-µl volume, which consists of 100 ng of genomic DNA from each sample, 10 µl of 10× Ex Taq buffer, 8 µl of a dNTP mixture (2.5 mM each dNTP), 5 µM each of ITS2F (5´-GAA TTA ATG TGA ACT GCA TAC TGC TT-3´) and ITS2R (5´-TTC CTC CGC TTA TTG ATA TGC TT-3´), and 0.5 µl of 5 U/µl of TaKaRa Ex Taq DNA Polymerase, using a TaKaRa PCR Thermal Cycler (Takara Bio Inc.). All PCR assays were performed using 1 cycle of 94˚C for 30 sec and 30 cycles of 98˚C for 10 sec, 60˚C for 30 sec, and 72˚C for 30 sec, followed by 1 cycle at 72˚C for 7 min and a final hold at 4˚C. Agarose gel electrophoresis (1.5%) with ethidium bromide staining was used to visualize the ITS2-PCR products.
Restriction fragment length polymorphism (RFLP) analysis of the ITS2 region
To evaluate interspecific restriction enzyme cutting site variation within the ITS2 region, this region was singled out for further study. The ITS2-PCR products were purified using a QIAquickTM PCR purification kit (Qiagen, Valencia, California, USA). To reveal the RFLP pattern, the purified PCR products were digested with Sau3A1 (Fermentas, Vilnius, Lithuania) according to the manufacturer’s instructions. The Sau3A1 reactions were incubated at 37˚C for 4 hr, at which point an additional 1 µl of Sau3A1 enzyme was added to each reaction. The reactions were then incubated overnight. The RFLP products were separated on 2% agarose gels, stained with ethidium bromide, photographed, and analyzed for fragment sizes.
Sequencing and analysis of the ITS2 region
The purified ITS2-PCR products were ultimately sequenced directly by SolGent (Daejeon, Korea) using the primers described previously. The complete ITS2 sequences for S. haematobium from 13 samples were compared to those isolated from Kenya (GenBank accession no. AF146038) using the Clone Manager software (Sci-Ed Software, Cary, North Carolina, USA).
Analysis of ITS-2, cox1, and nad1 genes of S. haematobium and S. mansoni using PCR
The mitochondrial cytochrome oxidase subunit 1(cox1), NADH dehydrogenase subunit 1 (nad1) and ITS-2 genes of S. haematobium and S. mansoni were analyzed using adult worms using PCR amplification. Briefly, genomic DNA was extracted from the adult worm using G-DEX™ genomic DNA extraction kit according to the manufacturer’s instructions. The primers used for PCR amplification were used the following primers: S. haematobium cox1 (DQ677664, 110 bp), forward 5´-AAA AGC TGT GGG TCT CGT GT-3´, reverse 5´-AAT GAA GAA GCG GAG AAA GC-3´; S. mansoni cox1 (NC_002545, 598 bp),forward 5´-TCA ATT TGA GAG GGG TCT GG-3´, reverse 5´-TGG GAT CTA AAA CCC GCA TA-3´; S. haematobium nad1 ((JQ595404,431 bp), forward 5´-GGC TGA TGT TCG TGA TCA AA-3´, reverse 5´-CGA AGT CGA GAA AAT GAA CCA-3´; S. mansoni nad1 (AF216698, 194 bp) forward 5´-TGG GGA GTG TGA GAG TGA GC-3´, reverse 5´-GAA AAG ACC CAC GCA TCA TT-3´; and S. mansoni ITS-2 (JQ289745, 104 bp) forward 5´-GAC GCA CAT TAA GTC GTG GA-3´, reverse 5´-CTG TGG CCG GAT TAT TAG GA-3´. Genomic DNA from S. haematobium eggs and S. mansoni adult worms were used for detection of specific genes. The PCR mixture for the PCR amplification contained 5 μl genomic DNA (200 ng/μl), 2 μl each of forward and reverse primers (10 μM), 4 μL dNTP (2.5 mM each), 5 μl 10× Ex Taq buffer, 0.25 μl Ex Taq polymerase (5 U/μl), and 31.75 μl DDW.PCR assays were performed with an initial denaturation step of 94˚C for 30 sec, followed by 30 cycles of denaturation at 98˚C for 10 sec, annealing at 56˚C for 30 sec, and extension at 72˚C for 60 sec, followed by 1 cycle at 72˚C for 10 min and a final hold at 4˚C.
Amplifications were generated using a TaKaRa PCR Thermal Cycler (Takara Bio Inc.). Agarose gel electrophoresis (1.5%) with ethidium bromide staining was used to visualize the cox1, nad1, and ITS2 of PCR products.
RESULTS
Results of multiplex PCR amplification
To select samples for the RFLP analysis of
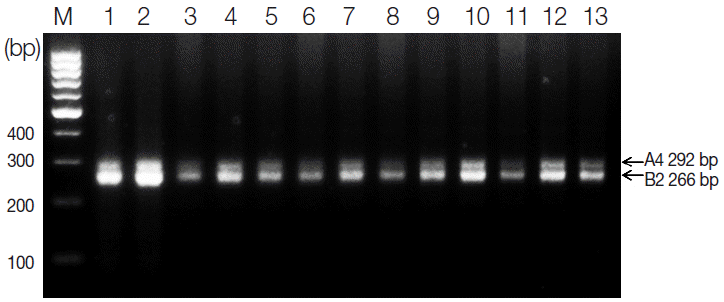
S. haematobium eggs from 73 Sudanese schoolchildren, multiplex PCR was performed using genomic DNA isolated from patients with microscopically confirmed urinary schistosomiasis. According to our multiplex PCR results, we selected 13 schistosomiasis-positive samples, each of which produced the expected 292-bp A4 and 266-bp B2 fragments (
Fig. 1). Also,
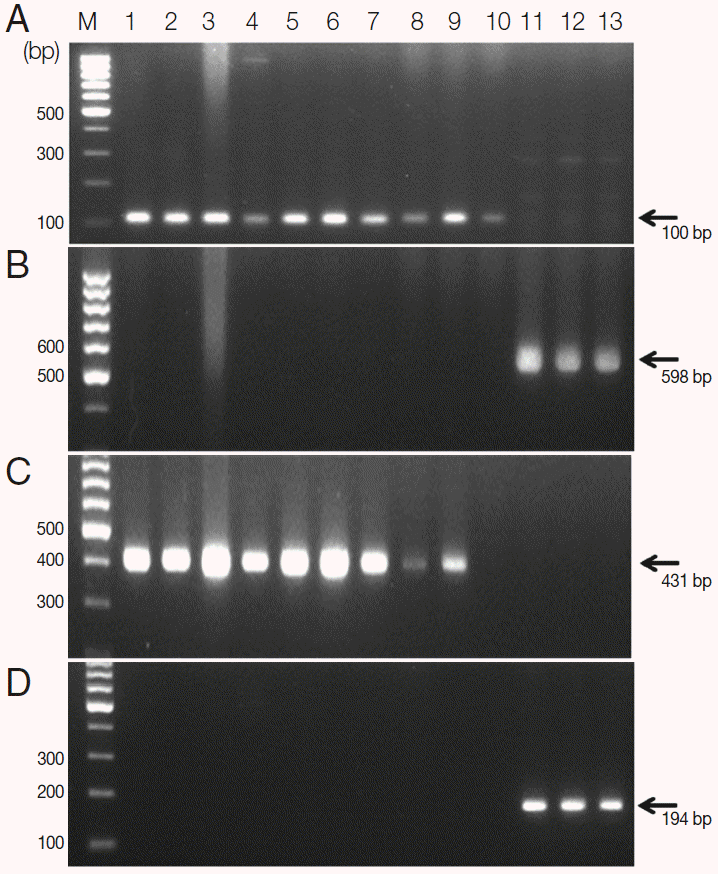
S. haematobium eggs were confirmed by PCR using
cox1 and
nad1 genes as the markers of genetic diversity. They showed species-specific
cox1 and
nad1 in comparison of those of
S. mansoni (
Fig. 2). These positive samples were used in the following genotyping study.
Using DNA from the 13 isolates collected in 5 areas of Sudan, PCR amplification using ITS2-specific primers produced 468-bp PCR product. Further, we examined species-specific variation in the enzyme cutting sites.
Sau3A1 cut the ITS2-containing PCR product of
S. haematobium in 3 places, producing fragments that were 237, 98, 83, and 50 bp in length (
Fig. 3). The number and location of these restriction enzyme sites were found to be consistent in both GenBank sequences isolated from Kenya. The selection of
Sau3A1 for our RFLP analysis was based on this lack of intraspecific variation in the presence and location of the restriction enzyme cutting sites. The RFLP patterns of the 13 specimens were similar, indicating that the
S. haematobium isolated from 5 different areas of Sudan were genetically identical in comparison to those collected in Kenya.
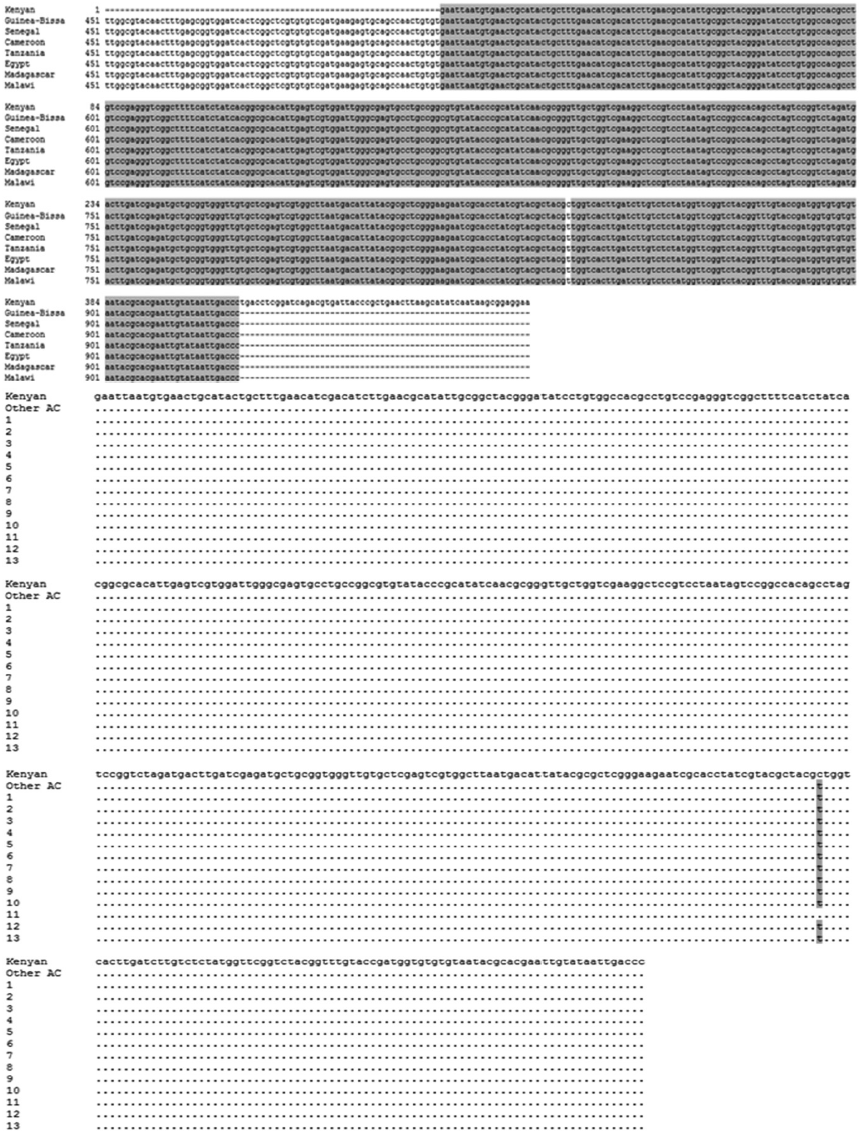
The 468-bp ITS2-PCR fragments were sequenced along both strands and compared to isolates from Kenya (AF146038, position 1-400) and other African countries including Guinea-Bissau (JQ397404), Senegal (FJ588861), Cameroon (JQ397406), Tanzania (GU257398), Egypt (JQ397407), Madagascar (JQ397414), and Malawi (JQ397410) (position 518-927). The ITS2 sequences of
S. haematobium obtained from other African countries including Guinea-Bissau, Senegal, Cameroon, Tanzania, Egypt, Madagascar, and Malawi area were 100% identical (data not shown). As shown in
Fig. 4, when the nucleotide sequences of the ITS2 region of
S. haematobium from 13 samples (position 1-400) obtained in 5 different areas of Sudan were compared to the ITS2 sequences obtained for
S. haematobium from Kenya (position 1-400) and other African countries (including Guinea-Bissau, Senegal, Cameroon, Tanzania, Egypt, Madagascar, and Malawi (position 518-927), there was only a single intraspecific base change at position 326 (‘T’ in 4 areas of Sudan) compared with Kenya sequence at position, while 100% was identical with those obtained from other African countries including Guinea-Bissau, Senegal, Cameroon, Tanzania, Egypt, Madagascar, and Malawi (position 518-927). The samples obtained from the Al Sidding area were 100% identical with
S. haematobium from Kenya; however, there was only a single intraspecific base change at position 326 (‘C’ ) compared with other African countries sequences at position 843 ‘T’.In addition, to compare the ITS2 gene between
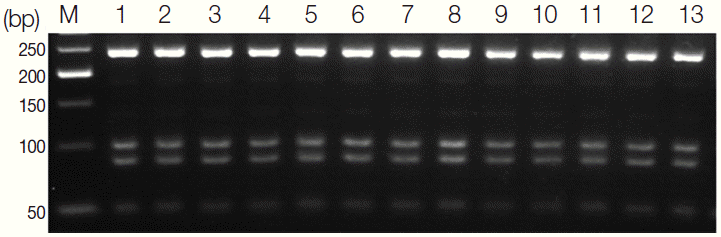
S. haematobium and S. mansoni, their ITS2 PCR fragments were sequenced. As shown in
Fig. 5, the nucleotide sequence of the PCR products from
S. mansoni were 99.0%, 99.0%, and 97.1% identical, respectively, to the ITS2 sequences of JQ289745 (position 619-722). However, ITS2 sequence of
S. haematobium (AF146038, position 112-215) was just 92.3% identical to the ITS2 sequences of
S. mansoni (JQ289745, position 619-722) (
Fig. 6). Thus, the ITS2 of
S. haematobium was different from that of S. mansoni.
DISCUSSION
The genetic diversity of human schistosome infections is influenced by a number of factors. Overlapping contact sites and snail and human movements may support the coexistence of a large number of genotypes in a given area. They can also engender the development of new parasitic strains or families through genetic interchange and recombination between local and introduced genotypes [
11]. Investigation of the population genetics of
S. haematobium is important for understanding its variable disease manifestations and epidemiology, and developing new strategies for treatment, vaccination, and diagnosis [
4,
5]. Dabo et al. [
12] found infections with multiple genotypes of
S. haematobium in the intermediate host of the organism (bulinid snails) using randomly amplified polymorphic DNA markers.
In this study, we first tested S. haematobium egg samples isolated from the urine of 73 infected children using the microsatellite markers A4 and B2 by PCR. A4 and B2 are useful for distinguishing S. haematobium from other Schistosoma species. However, our multiplex PCR results showed that 60 of the 73 samples did not produce the expected PCR band patterns for S. haematobium, suggesting that the number of S. haematobium eggs was insufficient for isolation of genomic DNA, and that the eggs were not from S. haematobium, or that polymorphisms in A4 and B2 exist among S. haematobium in Sudan (data not shown). Therefore, we grouped the samples according to their PCR band patterns and identified 13 samples that produced the expected PCR product size for markers A4 and B2; these were analyzed for genetic variation.
To determine the genotype of
S. haematobium within the final hosts, we examined the ITS2 region. Internal transcribed spacers are pieces of non-functional RNA situated between structural ribosomal RNAs on a common precursor transcript. ITS2 patterns can be used to rapidly differentiate
S. haematobium from
Schistosoma bovis by PCR-RFLP [
6]. However, the
Sau3A1-RFLP was not found in our selected groups, indicating that the eggs were indeed
S. haematobium. Next, to exclude the possibility of an unknown single nucleotide polymorphism in the ITS2 region, we sequenced the complete ITS2 region in our tested samples. According to our sequencing results, most of the A4- and B2-positive samples of
S. haematobium from Sudan were of a single origin and genotype that is dominant in African countries, including Guinea-Bissau, Senegal, Cameroon, Tanzania, Egypt, Madagascar, Mauritius Zambia, and Malawi, except for the sample from the Al Sidding region, which has experienced an inflow of Kenyan
S. haematobium. Although a limited number of
S. haematobium eggs with a stable genotype from 5 regions of Sudan were available, the finding of a single nucleotide alteration (326 C > T), which has been reported in Kenyan
S. haematobium, suggests that it is not a naturally occurring point mutation or polymorphism; instead, it suggests the inflow of Kenyan
S. haematobium into Sudan. Webster et al. [
13] have observed that according to DNA ‘barcoding’ study, using the
cox1 and
nad1 genes, low sequence variation was found among 41 localities representing 18 countries across Africa and the Indian Ocean Islands. Similarly, no obvious change in genetic diversity was detected on Zanzibar over a 4-year period [
14]. These studies provided evidence that if we analyze the genetic diversity using these mitochondrial genes of
cox1 and
nad1, there will not appear change in genotyping diversity.
According to geographic data, there is a direct connection between Lake Victoria in Kenya and Sudan (especially north Sudan) through the White Nile River, which passes through Uganda and South Sudan, suggesting that the spread of Kenyan
S. haematobium to Sudan might be based on water or a water-dwelling intermediate host of
Schistosoma. Monitoring of the abundance of Kenyan
S. haematobium along the White Nile River may help determine the inflow route of the strain. Another possible route for the inflow of Kenyan
Schistosoma to Sudan is by the migration of infected Kenyan people to Sudan. Whether the symptoms of schistosomiasis caused by the Kenyan strain of
S. haematobium differ in their severity compared to the strains in Sudan or other African countries requires further study; however, Brouwer et al. [
11] reported that the genetic diversity in parasite populations was considerable among children, and that this variability may impact acquired immunity and the clinical outcome of the infection.
Based on the results of genotyping with high-resolution and microsatellite markers, we found most of the S. haematobium population in Sudan consists of a pan-African S. haematobium genotype, and also we uncovered the possible inflow of a Kenyan strain of S. haematobium into Sudan. These data may serve as a baseline for future research, including population genetic analyses of S. haematobium in this region of Sudan.
Notes
-
We have no conflict of interest related to this work.
This study was done as the investigation for "The project for combating schistosomiasis in Sudan during 2012 through 2014" by Korea International Cooperation Agency (KOICA). We are indebted to several colleagues at Ministry of Health of Sudan and Embassy of the Republic of Korea in Sudan.
Fig. 1.Agarose gel electrophoresis of multiplex PCR products containing 2 microsatellite markers of Schistosoma haematobium (A4 and B2). Two bands were amplified from each sample: 292 bp for A4 and 266 bp for B4. M, 100-bp ladder. Lanes 1-2, Al Hidaeb; lanes 3-4, Al Zealet; lanes 5-6, Jazeera Aba; lanes 7-10, Khour Ajwal; lane 11, Al Sidding; and lanes 12-13, Al Zealet.
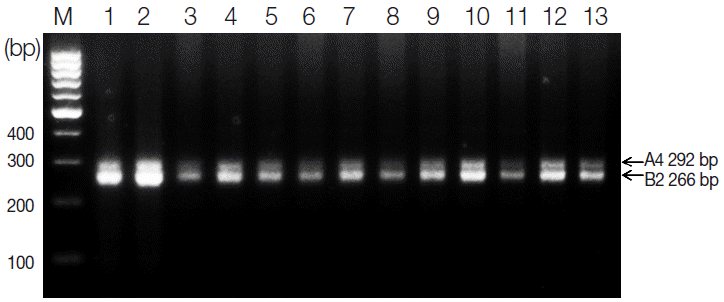
Fig. 2.Agarose gel electrophoresis of PCR products containing the mitochondrial cytochrome oxidase subunit 1 (cox1) and NADH dehydrogenase subunit 1 (nad1) of S. haematobium and S. mansoni. A, S. haematobium cox1 (DQ677664, 110 bp); B, S. mansoni cox1 (NC_002545, 598 bp); C, S. haematobium nad1 (JQ595404, 431 bp); D, S. mansoni nad1 (AF216698, 194 bp). M, 100 bp marker; lanes 1-10, S. haematobium eggs; lane 11-13, S. mansoni adult worms.
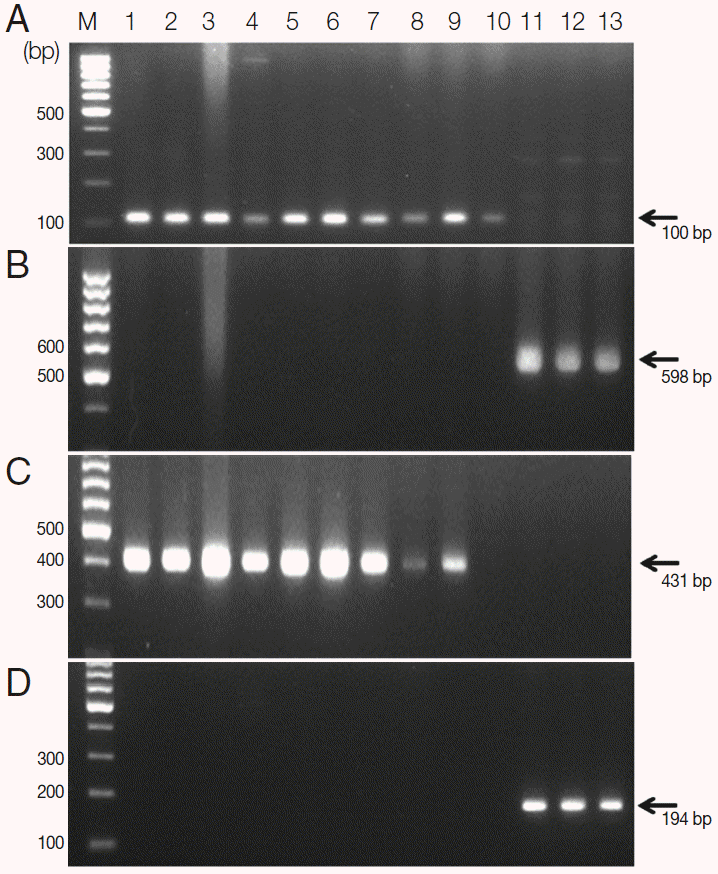
Fig. 3.Agarose gel stained with ethidium bromide showing the band patterns obtained by digestion of the 468-bp PCR product containing the second internal transcribed spacer (ITS2) with Sau3AI. Sau3A1 cut the ITS2-PCR product of S. haematobium in 3 places, producing visible fragments of 237, 98, 83, and 50 bp.
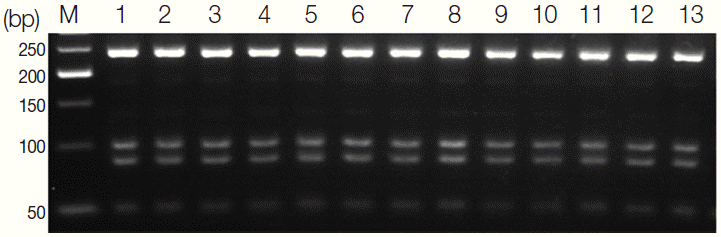
Fig. 4.The ITS2 nucleotide sequences of S. haematobium from 13 positive samples obtained from PCR products compared with a GenBank sequence isolated from Kenya (accession no. AF146038) and other African countries. Base homologies are indicated with a dot (·); base changes are shown in orange.
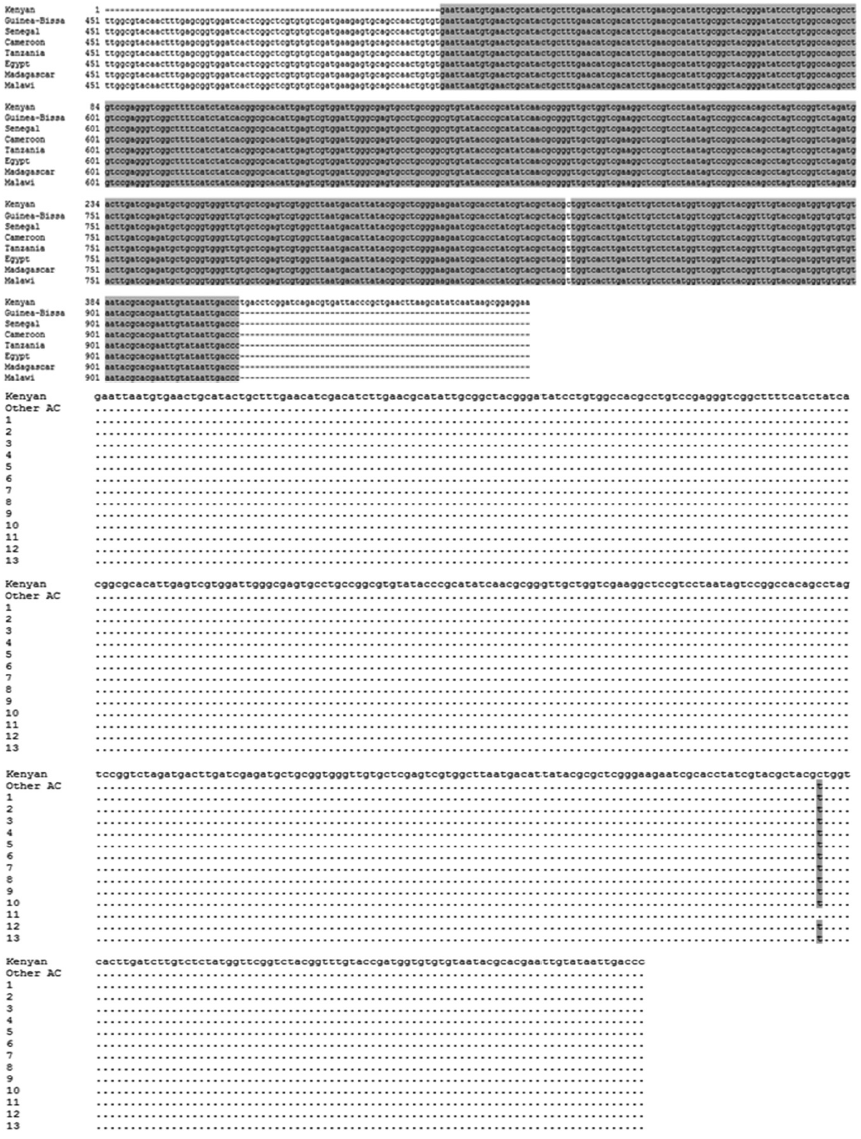
Fig. 5.The ITS2-PCR products and nucleotide sequences of S. mansoni adult worms. (A) Agarose gel electrophoresis of PCR products containing the ITS2 of the ribosomal gene complex from S. haematobium and S. mansoni. M, 100 bp marker; lane 1-10, S. haematobium eggs; lane 11-13: S. mansoni adult worms. (B) S. mansoni ITS2 nucleotide sequences of 3 positive samples obtained from PCR products compared with a GenBank sequence (accession no. JQ289745). The position of the S. mansoni ITS2 region was 619-722.

Fig. 6.The ITS2 nucleotide sequences of S. haematobium (AF146038, position 112-215) were compared with ITS2 sequences of S. mansoni (JQ289745, position 619-722). Base homologies are indicated with a dot (·); base changes are shown in orange.

References
- 1. Van der Werf MJ, de Vlas SJ, Brooker S, Looman CW, Nagelkerke NJ, Habbema JD, Engels D. Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. Acta Trop 2003;86:125-139.
- 2. Lee YH, Jeong HG, Kong WH, Lee SH, Cho HI, Nam HS, Ismail HA, Alla GN, Oh CH, Hong ST. Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. PLoS Negl Trop Dis 2015;86:e3423.
- 3. Ismail H, Hong ST, Babiker A, Hassan R, Sulaiman MA, Jeong HG, Kong WH, Lee SH, Cho HI, Nam HS, Oh CH, Lee YH. Prevalence, risk factors, and clinical manifestations of schistosomiasis among school children in the White Nile River basin, Sudan. Parasit Vectors 2014;7:478.
- 4. Norton AJ, Gower CM, Lamberton PH, Webster BL, Lwambo NJ, Blair L, Fenwick A, Webster JP. Genetic consequences of mass human chemotherapy for Schistosoma mansoni: population structure pre- and post-praziquantel treatment in Tanzania. Am J Trop Med Hyg 2010;83:951-957.
- 5. Gower CM, Gabrielli AF, Sacko M, Dembelé R, Golan R, Emery AM, Rollinson D, Webster JP. Population genetics of Schistosoma haematobium: development of novel microsatellite markers and their application to schistosomiasis control in Mali. Parasitology 2011;138:978-994.
- 6. Barber KE, Mkoji GM, Loker ES. PCR-RFLP analysis of the ITS2 region to identify Schistosoma haematobium and S. bovis from Kenya. Am J Trop Med Hyg 2000;62:434-440.
- 7. Zhao QP, Jiang MS, Littlewood DT, Nie P. Distinct genetic diversity of Oncomelania hupensis, intermediate host of Schistosoma japonicum in mainland China as revealed by ITS sequences. PLoS Negl Trop Dis 2010;4:e611.
- 8. Ezeh C, Yin M, Li H, Zhang T, Xu B, Sacko M, Feng Z, Hu W. High genetic variability of Schistosoma haematobium in Mali and Nigeria. Korean J Parasitol 2015;53:129-134.
- 9. Van den Broeck F, Geldof S, Polman K, Volckaert FA, Huyse T. Optimal sample storage and extraction procotols for reliable multilocus genotyping of the human parasite Schistosoma mansoni. Infect Genet Evol 2011;11:1413-1418.
- 10. Golan R, Gower CM, Emery AM, Rollinson D, Webster JP. Isolation and characterization of the first polymorphic microsatellite markers for Schistosoma haematobium and their application in multiplex reactions of larval stages. Mol Ecol Resour 2008;8:647-649.
- 11. Brouwer KC, Ndhlovu P, Munatsi A, Shiff CJ. Genetic diversity of a population of Schistosoma haematobium derived from schoolchildren in east central Zimbabwe. Parasitology 2001;87:762-769.
- 12. Dabo A, Durand P, Morand S, Diakite M, Langand J, Imbert-Establet D, Doumbo O, Jourdane J. Distribution and genetic diversity of Schistosoma haematobium within its bulinid intermediate hosts in Mali. Acta Trop 1997;66:15-26.
- 13. Webster BL, Emery AM, Webster JP, Gouvras A, Garba A, Diaw O, Seye MM, Tchuente LA, Simoonga C, Mwanga J, Lange C, Kariuki C, Mohammed KA, Stothard JR, Rollinson D. Genetic diversity within Schistosoma haematobium: DNA barcoding reveals two distinct groups. PLoS Negl Trop Dis 2012;6:e1882.
- 14. Stothard JR, Ameri H, Khamis IS, Blair L, Nyandindi US, Kane RA, Johnston DA, Webster BL, Rollinson D. Parasitological and malacological surveys reveal urogenital schistosomiasis on Mafia Island, Tanzania to be an imported infection. Acta Trop 2013;128:326-333.