Abstract
The 2 species of the genus Anoplocephala (Anoplocephalidae), A. perfoliata and A. magna, are among the most important equine cestode parasites. However, there is little information about their differences at the molecular level. The present study revealed that the mitochondrial (mt) genome of A. magna was 13,759 bp in size and 700 bp shorter than that of A. perfoliata. The 2 species includes 2 rRNA, 22 tRNA, and 12 protein-coding genes each. The size of each of the 36 genes was the same as that of A. perfoliata, except for cox1, rrnL, trnC, trnS2(UCN), trnG, trnH, trnQ, and trnP. In the full mitochondrial genome, the sequence similarity was 87.1%. The divergence in the nucleotide and amino acid sequences of individual protein-coding genes ranged from 11.1% to 16% and 6.8% to 16.4%, respectively. The 2 noncoding regions of the mt genome of A. magna were 199 bp and 271 bp in length, while the equivalent regions in A. perfoliata were 875 bp and 276 bp, respectively. The results of this study support the proposal that A. magna and A. perfoliata are separate species, consistent with previous morphological analyses.
-
Key words: Anoplocephala magna, Anoplocephala perfoliata, mitochondrial genome
Equine tapeworm infections are caused by species of the family
Anoplocephalidae in the order Cyclophyllidea. The
Anoplocephalidae family has 2 valid genera,
Anoplocephala (
A. perfoliata and
A. magna) and
Anoplocephaloides (
A. mamillana). Previous studies have shown that both
A. perfoliata and
A. magna have high prevalence in the equine population, and in many cases occur together as a mixed infection [
1]. These tapeworms are found in and around the ileocecal valve of horses and are thought to be associated with several intestinal diseases [
2,
3] that lead to reduced body weight of horses. To minimize economic loss, accurate identification and differentiation is needed to help control these equine parasites. Additionally, the genetics, epidemiology, and biology of the species (
Anoplocephalidae) are as yet poorly understood.
Complete mitochondrial DNAs have been used effectively to analyze species phylogenetics, ecology, and population genetics, and some genes and gene regions have helped us locate novel molecular markers [
2-
6]. The complete mitochondrial DNA of platyhelminthes comprises 12 protein-coding genes, 2 ribosomal genes and 22 transfer RNA genes [
5]. The complete mitochondrial genome of
A. perfoliata was recently sequenced; however, that of
A. magna remains undetermined.
In the present study, the complete mitochondrial genome of A. magna was sequenced and compared with that of A. perfoliata of the genus Anoplocephala in order to find useful molecular markers for the identification of the 2 most commonly occurring equine parasites. Determining the complete mitochondrial genome of A. magna will provide new molecular data for future studies of comparative mitochondrial genomics as well as the phylogenetics of parasitic cestodes.
Equine tapeworms of the species were collected from the digestive tracts of donkeys slaughtered at a commercial slaughterhouse in China and identified by morphology. Total DNA was extracted from a single sample, A. magna, with a Miniprep DNA extraction kit (AXYGEN, Avenue Union City, California, USA).
The complete mt genomic sequence of
A. magna was amplified in 3 overlapping fragments. The 3 pair-conserved primers and amplifying conditions used for the long-PCR reactions were the same as those described for amplifying corresponding fragments of
A. perfoliata [
7]. These amplicons were sequenced by Shanghai Sangon (Shanghai, China) using primer-walking in both directions.
The complete mt genomic sequence of
A. magna was assembled using CAP3 Program [
8] and annotated using ClustalX software based on comparison sequence with that of
A. perfoliata reported previously [
7]. The gene boundaries of the mt genomic sequence of
A. magna were identified by alignment in comparison to those of
A. perfoliata. Total 22 tRNA genes were found using ARWEN [
9] and further determined by checking anticodon sequences and potential secondary structures. Putative stem-loop structures of non-coding regions were inferred through comparison with similar published sequences. The amino acid sequences of 12 protein-coding genes were deduced using the genetic code set for the flatworm (Translation Table 9). Nucleotide and amino acid sequence differences between
A. magna and
A. perfoliata were established with pairwise comparisons.
The complete circular mt genome of
A. magna was 13,759 bp in size (GenBank accession no. KU236385).
A. magna contains 36 genes, including 12 protein-coding genes (
cox1-3,
atp6,
nad1-6,
nad4L, and
cytb), 22 transfer RNA genes (
trn), and 2 ribosomal RNA genes (the small and large subunits of rRNA were designed as
rrnS and
rrnL) (
Table 1). Putative gene arrangement and lengths of
A. magna are listed in
Table 1. All 36 genes are transcribed in the same direction. Gene overlaps were found between
nad4 and
nad4L, between
trnQ and
trnF, between
trnF and
trnM, and between
trnV and
trnA, as reported in
A. perfoliata [
7]. The nucleotide composition of
A. magna mtDNA is biased toward T and A (70.8%), as observed with
A. perfoliata (71.0%) and other flatworm mt genomes [
2-
6]. The genome organization and structure of
A. magna is the same as that of
A. perfoliata (
Table 1).
Twelve protein-coding genes in the
A. magna mtDNA comprised 73.3% of the total length. The proportion was similar to data for other cestodes [
2-
6], but higher than that of
A. perfoliata (69.8%). This difference in proportion between
A. magna and
A. perfoliata was caused by the length variation of the non-coding region (
Table 1). Eight of the 12 protein-coding genes used ATG as their initiation codon, and 4 used GTG. Eleven genes had a complete stop codon with 4 genes using TAA and 7 genes using TAG (
Table 1). One gene,
cox3, was predicted to end with an incomplete termination codon, as identified in the
cox3 of
A. perfoliata [
7]. The size of each of the 12 protein-coding genes identified in the mt genome of
A. magna was the same as that of
A. perfoliata, except for the
cox1 gene whose size was 1,590 bp in
A. magna and 1,593 bp in
A. perfoliata.
A total of 22 tRNAs were identified in
A. magna, of which 18 were inferred to have their nucleotide sequences folded into conventional secondary structures. Meanwhile, the other 4 tRNAs (
trnS1,
trnS2,
trnC, and
trnR) were predicted to have lost the DHU arms, as in the case of
A. perfoliata. In
A. magna, the size of individual tRNA genes ranged from 57 bp to 71 bp, and the size of 5 tRNA genes in its mt genome varied compared to those of the corresponding genes in
A. perfoliata (
Table 1), including
trnC (3 bp),
trnS2 (2 bp),
trnG (1 bp),
trnQ (1 bp), and
trnP (1 bp). Among the predicated 36 genes for the
A. magna mt genome, only the
trnF gene sequence was identical to that of
A. perfoliata.
In the mt genome of A. magna, rrnS was located between trnC and cox2 and was 724 bp in size. The rrnL was located between trnT and trnC, and was 973 bp in size. The respective sizes of rrnS and rrnL in A. perfoliata were 724 bp and 981 bp.
A total of 22 non-coding regions, ranging from 1 bp to 271 bp in size, were found in the
A. magna mt genome (
Table 1). The 2 largest non-coding regions were named as NC1 and NC2. NC1 was located between
trnY and
trnS2 (UCN), and NC2 was located between
nad5 and
trnG. The lengths of NC1 and NC2 were 199 bp and 274 bp, respectively, for
A. magna, and 875 bp and 279 bp for
A. perfoliata.
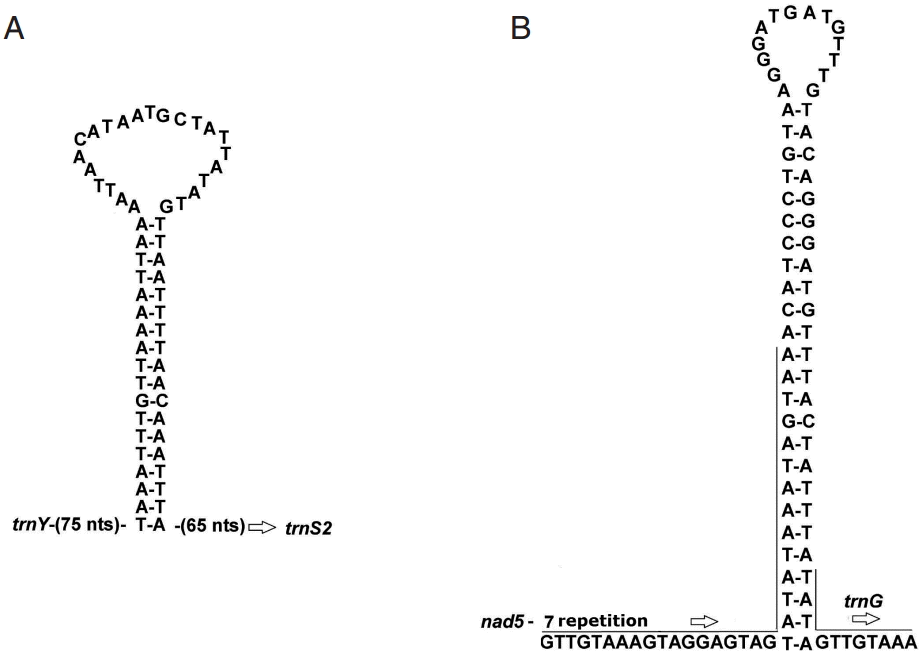
The possible secondary structures for the 2 largest non-coding regions are shown in
Fig. 1. The partial sequence in the NC1 of
A. magna could form a significant stable stem-loop structure with 18 canonical base pairs and a loop of 23 nt (
Fig. 1A). NC2 comprises 7 identical repeats of a 32 nt sequence, and part of this 7th repeat and the remaining 50 bp can fold a perfectly matched stem-loop structure with 25 base pairs and a loop of 14 nt (
Fig. 1B).
The overall difference in nucleotide sequence between
A. magna and
A. perfoliata was 12.9%. The nucleotide sequence divergence for
rrnS and
rrnL was 8.2% and 9.2%, respectively, revealing that
rrnS is more conserved than
rrnL. The sequence difference in NC1 between the 2 species was 24.1%, and that of NC2 was 7.7%. The divergence in the nucleotide and amino acid sequences of each of the 12 mt proteins in
A. magna and
A. perfoliata ranged from 11.1% to 16% and 6.8% to 16.4%, respectively (
Table 1). The amino acid sequence of
nad4L gene was the least conserved protein, and that of
cox2 was the most conserved protein.
The lengths of the complete genomes of
A. perfoliata and
A. magna were 14,459 bp and 13,759 bp, respectively. The difference between the lengths of the complete genomes was largely due to differences in the length of NC1, which is caused by the differential number of identical 169-nucleotide repetitive sequence units. Compared to the NC1 of
A. magna, additional 4-repetitive sequence units were found in that of
A. perfoliata. The difference in the number of repetitive sequence units in this non-coding region was similar to that between
Diphyllobothrium latum and
D. nihonkaiense [
10]. The significance of the number variation of repetitive sequence units between closely-related species needs to be evaluated further.
Stem loop structure has been predicted in the non-coding regions in
A. magna mtDNA, as in the mtDNAs of many cestodes [
6] and in nematode mtDNAs [
11]. Non-coding regions of the
A. magna mt genome which have stem-loop structures similar to those in vertebrates and invertebrates may play a similar function to those found in vertebrates, which are known to be involved in the processes of replication and transcription [
12-
14].
A substantial difference (12.9%) in the nucleotide sequences was observed between the complete mt genomes of
A. magna and
A. perfoliata from China. The finding of the sequence variation in the mt genome between
A. magna and
A. perfoliata was consistent with a previous study, where nucleotide sequence variation was detected in the nuclear ITS rDNA [
1]. In this study, the differences in the nucleotide and amino acid sequences between
A. magna and
A. perfoliata support the hypothesis that they are 2 distinct species. To date, only a molecular marker of internal transcribed spacer 2 has been proposed to be used for differentiating infections caused by the 2 most frequently encountered equine tapeworms,
A. magna and
A. perfoliata [
1]. In this study, more gene regions were found to have greater interspecific variation including
nad5,
cytb,
nad4,
atp6,
nad6,
nad3,
nad1, and
cox3 and may thus be considered as ecological and diagnostic markers for the 2 species. Actually, some mt genes (
cox1 and
cytb) have been used as targets for molecular-based methods to identify species in other cestodes [
15-
18].
This study provided new and valuable information about the mt genome of A. magna. The mt genome data presented here supports the hypothesis that A. magna and A. perfoliata represent distinct species. The complete mt genome of A. magna can now be used for comparative mitogenomics among members of the family Anoplocephalidae to find the best molecular markers for characterization and will also be used to reconstruct the molecular systematics of the order Cyclophyllidea in the future.
Notes
-
There is no conflict of interest related to this work.
The project support was provided by the International Science & Technology Cooperation Program of China (grant no. 2013DFA31840).
Fig. 1.Putative secondary structure of the 2 non-coding regions NC1 and NC2 of Anoplocephala magna. NC1 (A) located between trnY and trnS2, and NC2 (B) between nad5 and trnG.
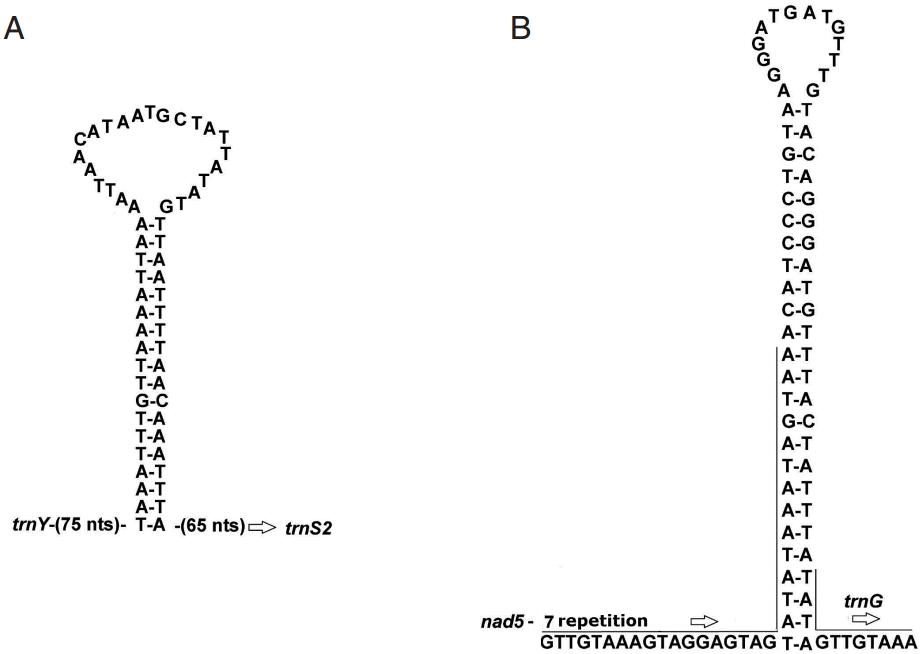
Table 1.Organization and comparison of mitochondrial genomes of Anoplocephala magna (Am) and Anoplocephala perfoliata (Ap)
Table 1.
|
Genes |
Start and end points/length (nt)
|
Divergence (%) Ap/Am
|
|
Ap |
Am |
Nucleotide |
Polypeptide |
|
cox1
|
1-1593/1593 |
1-1590/1590 |
11.6 |
7.8 |
|
tRNA-Thr (T) |
1596-1659/64 |
1593-1656/64 |
6.2 |
|
|
rrnL
|
1660-2640/981 |
1657-2629/973 |
9.2 |
|
|
tRNA-Cys (C) |
2641-2706/66 |
2630-2692/63 |
9.5 |
|
|
rrnS
|
2707-3430/724 |
2693-3416/724 |
8.2 |
|
|
cox2
|
3431-4006/576 |
3417-3992/576 |
11.1 |
6.8 |
|
tRNA-Glu (E) |
4008-4071/64 |
3993-4056/64 |
6.2 |
|
|
nad6
|
4076-4534/459 |
4061-4519/459 |
14.8 |
15.1 |
|
tRNA-Tyr (Y) |
4541-4606/66 |
4522-4587/66 |
12.1 |
|
|
Non-coding region (NC1) |
4607-5481/875 |
4588-4786/199 |
24.1 |
|
|
tRNA-SerUCN (S2) |
5482-5554/73 |
4787-4857/71 |
9.9 |
|
|
tRNA-LeuCUN (L1) |
5582-5642/61 |
4873-4933/61 |
6.6 |
|
|
tRNA-LeuUUR (L2) |
5654-5717/64 |
4946-5009/64 |
10.9 |
|
|
tRNA-Arg (R) |
5727-5783/57 |
5018-5074/57 |
8.9 |
|
|
nad5
|
5785-7365/1581 |
5076-6656/1581 |
16.0 |
14.9 |
|
Non-coding region (NC2) |
7366-7644/279 |
6657-6930/274 |
7.7 |
|
|
tRNA-Gly (G) |
7642-7704/63 |
6928-6991/64 |
6.3 |
|
|
cox3
|
7708-8351/644 |
6995-7638/644 |
14.8 |
14.0 |
|
tRNA-His (H) |
8352-8418/67 |
7639-7705/67 |
3.0 |
|
|
cytb
|
8422-9522/1101 |
7709-8809/1101 |
15.1 |
14.5 |
|
nad4L
|
9525-9785/261 |
8811-9071/261 |
13.0 |
16.3 |
|
nad4
|
9752-10999/1248 |
9038-10285/1248 |
15.1 |
11.3 |
|
tRNA-Gln (Q) |
11001-11063/63 |
10287-10350/64 |
7.9 |
|
|
tRNA-Phe (F) |
11062-11123/62 |
10349-10410/62 |
0.0 |
|
|
tRNA-Met (M) |
11120-11184/65 |
10407-10471/65 |
6.2 |
|
|
atp6
|
11188-11703/516 |
10475-10990/516 |
14.9 |
10.5 |
|
nad2
|
11718-12593/876 |
11004-11879/876 |
11.5 |
11.7 |
|
tRNA-Val (V) |
12598-12660/63 |
11899-11961/63 |
4.8 |
|
|
tRNA-Ala (A) |
12660-12724/65 |
11961-12025/65 |
6.2 |
|
|
tRNA-Asp (D) |
12726-12789/64 |
12027-12090/64 |
4.7 |
|
|
nad1
|
12793-13683/891 |
12094-12984/891 |
14.7 |
10.5 |
|
tRNA-Asn (N) |
13710-13778/69 |
13008-13076/69 |
8.7 |
|
|
tRNA-Pro (P) |
13784-13849/66 |
13083-13149/67 |
9.1 |
|
|
tRNA-Ile (I) |
13850-13912/63 |
13150-13212/63 |
4.8 |
|
|
tRNA-Lys (K) |
13917-13978/62 |
13219-13280/62 |
4.8 |
|
|
nad3
|
13983-14330/348 |
13285-13632/348 |
14.7 |
16.5 |
|
tRNA-SerAGN (S1) |
14331-14390/60 |
13633-13692/60 |
15.0 |
- |
|
tRNA-Trp (W) |
14393-14454/62 |
13693-13754/62 |
1.6 |
- |
References
- 1. Bohorquez GA, Luzon M, Martin-Hernandez R, Meana A. New multiplex PCR method for the simultaneous diagnosis of the three known species of equine tapeworm. Vet Parasitol 2015;207:56-63.
- 2. Jeon HK, Kim KH, Eom KS. Complete sequence of the mitochondrial genome of Taenia saginata: comparison with T. solium and T. asiatica. Parasitol Int 2007;56:243-246.
- 3. Jia WZ, Yan HB, Guo AJ, Zhu XQ, Wang YC, Shi WG, Chen HT, Zhan F, Zhang SH, Fu BQ, Littlewood DTK, Cai XP. Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genomics 2010;11:447.
- 4. Nakao M, Lavikainen A, Iwaki T, Haukisalmi V, Konyaev S, Oku Y, Okamoto M, Ito A. Molecular phylogeny of the genus Taenia (Cestoda: Taeniidae): proposals for the resurrection of Hydatigera Lamarck, 1816 and the creation of a new genus Versteria. Int J Parasitol 2013;43:427-437.
- 5. Littlewood DT, Lockyer AE, Webster BL, Johnston DA, Le TH. The complete mitochondrial genomes of Schistosoma haematobium and Schistosoma spindale and the evolutionary history of mitochondrial genome changes among parasitic flatworms. Mol Phylogenet Evol 2006;39:452-467.
- 6. von Nickisch-Rosenegk M, Brown WM, Boore JL. Complete sequence of the mitochondrial genome of the tapeworm Hymenolepis diminuta: gene arrangements indicate that Platyhelminths are Eutrochozoans. Mol Biol Evol 2001;18:721-730.
- 7. Guo A. The complete mitochondrial genome of Anoplocephala perfoliata, the first representative for the family Anoplocephalidae. Parasit Vectors 2015;8:549.
- 8. Huang X, Madan A. CAP3: a DNA sequence assembly program. Genome Res 1999;9:868-877.
- 9. Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008;24:172-175.
- 10. Yamasaki H, Ohmae H, Kuramochi T. Complete mitochondrial genomes of Diplogonoporus balaenopterae and Diplogonoporus grandis (Cestoda: Diphyllobothriidae) and clarification of their taxonomic relationships. Parasitol Int 2012;61:260-266.
- 11. Okimoto R, Chamberlin HM, Macfarlane JL, Wolstenholme DR. Repeated sequence sets in mitochondrial DNA molecules of root knot nematodes (Meloidogyne): nucleotide sequences, genome location and potential for host-race identification. Nucleic Acids Res 1991;19:1619-1626.
- 12. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res 1999;27:1767-1780.
- 13. Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol 1992;141:173-216.
- 14. Monnerot M, Solignac M, Wolstenholme DR. Discrepancy in divergence of the mitochondrial and nuclear genomes of Drosophila teissieri and Drosophila yakuba. J Mol Evol 1990;30:500-508.
- 15. Nkouawa A, Sako Y, Nakao M, Nakaya K, Ito A. Loop-mediated isothermal amplification method for differentiation and rapid detection of Taenia species. J Clin Microbiol 2009;47:168-174.
- 16. McManus DP. Molecular discrimination of taeniid cestodes. Parasitol Int 2006;55(suppl):S31-S37.
- 17. Yamasaki H, Allan JC, Sato MO, Nakao M, Sako Y, Nakaya K, Qiu D, Mamuti W, Craig PS, Ito A. DNA differential diagnosis of taeniasis and cysticercosis by multiplex PCR. J Clin Microbiol 2004;42:548-553.
- 18. Gonzalez LM, Montero E, Morakote N, Puente S, Diaz De Tuesta JL, Serra T, López-Velez R, McManus DP, Harrison LJ, Parkhouse RM, Gárate T. Differential diagnosis of Taenia saginata and Taenia saginata asiatica taeniasis through PCR. Diagn Microbiol Infect Dis 2004;49:183-188.